Institutional Review Board (IRB) Guidebook
This guidebook includes all of the information previously found on the Institutional Review Board (IRB) webpage.
After clicking into a topic, navigation within the book or page can be found on the left side of your screen. Above the page lists your current location within the content structure. If needed you will find a search function at the top of the page which may also be used for fast navigation.
The FAQ page will assist in directing you to the appropriate section of the guidebook based on your question.
If you have any questions, please contact us at 402-559-6463 or by email at irbora@unmc.edu
- Table of Contents
- Introduction
- IRB News & Updates
- Frequently Asked Questions (FAQ)
- HRPP (Human Research Policy & Procedures) Manual
- Does my project require IRB review?
- Which application do I use?
- Submission deadlines
- Report a research problem or complaint
- Procedures
- Adverse Event Reporting
- ClinicalTrials.gov (CT.gov)
- Emergency Treatment
- Exempt Studies
- Incidents (Non-compliance/Problems)
- Protocol Deviations
- Submission Process
- Education & Resources
- HRPP Investigator Guidance Series
- Investigator Resources
- IRB Conference Content
- Mental health considerations
- Miscellaneous Resources
- Training
- CITI Training
- Community Partners
- Consent Forms
- E-Signature Instructions
- IRB Training Videos
- RSS Training
- Virtual Training/Office Hours
- Forms
- Existing paper protocols
- FWAs - Federal Wide Assurance
- Miscellaneous
- Recruitment templates
- Short Forms
- Subject's rights & responsibilities
- Single & Central IRB
- Glossary
- sIRB - What is Single IRB?
- sIRB - UNMC process
- sIRB - Submission deadlines
- sIRB - Reliance
- sIRB - Fees
- cIRB - What is Central IRB?
- cIRB - UNMC Process
- cIRB - Reliance
- cIRB - Fees
- cIRB - Forms & Links
- SROC
- IRB Staff
Table of Contents
Does my project require IRB review?
Report a research problem or complaint
- Adverse Event Reporting
- Clinicaltrials.gov
- Emergency Treatment
- Exempt Studies
- Incidents (Non-compliance/Problems)
- Protocol Deviations
- Submission Process
- HRPP Investigator Guidance Series
- Investigator Resources
- IRB Conference Content
- Mental Health Considerations
- Miscellaneous Resources
- CITI Training
- Community Partners
- Consent Forms
- E-Signature Instructions
- IRB Training Videos
- RSS Training
- Virtual Training/Office Hours
- Existing Paper Protocols
- FWAs - Federal Wide Assurance
- Miscellaneous
- Recruitment templates
- Short Forms
- Subject's Rights & Responsibilities
- Glossary
- sIRB - What is Single IRB?
- sIRB - UNMC process
- sIRB - Submission deadlines
- sIRB - Reliance
- sIRB - Fees
- cIRB - What is Central IRB?
- cIRB - UNMC Process
- cIRB - Reliance
- cIRB - Fees
- cIRB - Forms & Links
Introduction
 Mailing Address:
Mailing Address:
| Institutional Review Board |
|---|
| University of Nebraska Medical Center |
| 987830 Nebraska Medical Center |
| Omaha, NE 68198-7830 |
| Phone: 402-559-6463 Email: irbora@unmc.edu |
The Institutional Review Board (IRB) is a committee formally constituted under federal regulations and institutional policy, and charged with reviewing research involving human subjects to protect the rights and welfare of those subjects. The IRB is composed of members from a variety of scientific disciplines as well as persons from the community. The IRB has the authority to approve, require modifications in (to secure approval), or disapprove research. The IRB also serves as a resource for researchers by providing advice and guidance on ethical and regulatory issues related to human subject research.
The Office of Regulatory Affairs (ORA) is the administrative department that provides support for the functions of the IRB. The ORA also serves as a final gateway for research review by assuring that all other institutional committee reviews and approvals have been obtained, and that all institutional requirements have been met.
The IRB and the ORA are committed to facilitating the conduct of ethical research involving human subjects, in full compliance with federal, state and institutional requirements.
Please contact us at irbora@unmc.edu for assistance with the submission of all IRB Applications and forms.
The pages and chapters to the right of your screen will take you to relevant topics and areas of interest. If there is anything you would like to see added to this guidebook, please let us know.
If you are new to research and working with the IRB, please visit this page for an introduction to the regulatory process.
Information regarding IRB staff, chairs, IRB members, and the Assistant Vice Chancellor; as well as contact information can be found here.
IRB Office Hours
We can be reached by phone (402-559-6463) or by email (irbora@unmc.edu) Mon-Fri 7am-5pm.
The IRB offers two virtual learning/training sessions each month.
Please direct questions regarding virtual training hours to the IRB Education Coordinator: Megan Berger mberger@unmc.edu or 402-559-6044
IORG #0000392
UNMC IRB Federal Wide Assurance
Adult IRB Roster -2025 (FWA00002939)
Pediatric IRB Roster -2025 (FWA00002939)
IRB Number for Each IRB:
- IRB-01 (Adult; first Thursday of every month except January and July)
- IRB00000670
- IRB-02 (Adult; third Thursday of every month)
- IRB00000671
- IRB-03 (Rapid Response)
- IRB00002686
- IRB-04 (Pediatric; fourth Tuesday of every month)
- IRB00007222
- IRB-05 (SIRB; second Friday of every month)
- IRB00012770



IRB News & Updates
Congruency, Consent, and Recruitment Materials
Dec 13th, 2024
In order to assure that subject injury language in the consent forms matches that in contracts, UNeHealth or SPA will begin conducting congruency checks. The congruency check is an institutional requirement and is a requirement for accreditation of our Human Research Protection Program.
Research teams should ensure a copy of the consent form is either developed or uploaded in RSS. The Office of Regulatory Affairs will notify SPA/UNeHealth of the requirement for a congruency check. If a discrepancy is found, SPA/UNeHealth will reach out to study teams directly to correct the consent form. Research teams will need to revise the consent developed in RSS or upload a copy of the revised consent (if relying on an external IRB) to RSS.
Congruency checks apply to studies that are conducted at UNMC, Nebraska Medicine, and Children's Nebraska that are:
- Commercially funded
- Grant/Foundation/Consortium (other than NIH) funded
- DoD funded
Congruency checks are NOT required for:
- NIH funded studies
- Cooperative Groups studies, such as the Children's Oncology Group
- Departmentally funded studies
- CCTR funded studies
- Studies conducted at UNO
CIRB Consent Forms
For studies relying on an external IRB for oversight (CIRB studies), the Office of Regulatory Affairs will be reviewing consent forms to verify the required UNMC local consent language has been appropriately inserted into the consent. The required language is available on the UNMC IRB website. This review will also look for any consent language required by the UNMC Conflict of Interest Committee, as applicable.
CIRB Recruitment Materials
For studies relying on an external IRB for oversight (CIRB studies), the Office of Regulatory Affairs will begin reviewing recruitment to verify the materials conform to UNMC HRPP policies 3.5 and 3.6 . For existing studies, any new recruitment materials should be submitted as a change request. For new studies, recruitment materials should be uploaded to RSS at the time of initial submission.
Fee Increase
July 31st, 2024
To align with other academic medical centers and universities, effective September 1, 2024, the Office of Regulatory Affairs and the IRB will increase the IRB review fees for the initial review of protocols where UNMC relies on a commercial IRB. This will apply only to protocols submitted on or after September 1, 2024.
- Central IRB initial review fee (UNMC relying on a commercial IRB) will increase from $1500 to $3000. This is a one-time fee.
All investigators involved in research projects relying on a commercial IRB should include the IRB review fee in their grant or contract budget. A funding account must be provided at the time of IRB submission.
If you have any questions, please contact the Office of Regulatory Affairs at irbora@unmc.edu.
Advarra Submissions
July 2nd, 2024
Effective immediately, UNMC site submissions to Advarra may begin as soon as a study has been assigned a UNMC IRB number in the RSS system. Previously, Advarra required an Acceptance letter from the Office of Regulatory Affairs in order for study teams to begin the submission process with Advarra. Study teams will be required to provide Advarra with a copy of the "New Protocol" email as documentation of having a UNMC IRB number.
Emergency Preparedness (EP/COOP)
May 16th, 2024
We would like to remind clinical researchers about our Emergency Preparedness & Continuity of Operations Plan (EP/COOP), which is designed to:
- Detail how decisions on altering or halting ongoing research might affect studies, investigators, and current/potential subjects.
- Establish a framework to restore essential functions to UNMC’s Human Research Protection Program (HRPP), Institutional Review Board (IRB), and ORA during emergencies.
- Complement the broader UNMC/NM enterprise-wide COOP.
The plan is available here: https://guides.unmc.edu/books/hrpp-policies-and-procedures/page/emergency-preparedness-continuity-of-operations-plan-%28epcoop%29
IRB Office Hours
January 19th, 2024
Intended for questions regarding research projects, filling in an application, policies, regulations, etc.
-
2nd Monday of every month 9-10 AM (Zoom link)
-
4th Thursday of every month 2-3 PM (Zoom link)
Please direct questions regarding office hours to the IRB Education Coordinator, Megan Berger: mberger@unmc.edu or 402-559-6044
We would like to remind everyone that IRB staff are available for assistance outside of office hours. If you ever need help outside of the designated office hours, please do not hesitate to reach out to an analyst or our main email, IRBORA@unmc.edu. We are happy to assist you via email, phone call, or video conference call during normal work hours.
Additional information can be found on our Education page [here](insert link here).
August 17th, 2023
The Office of the Vice Chancellor for Research and Office of Regulatory Affairs want to inform human subjects researchers about a temporary policy change.
Why it Matters: Over the next couple of months, temporary onboarding and training demands for new IRBORA staff may lead to a delay in timelines for IRB review and other ORA processes. The VCR and ORA recognize the potential impacts this could have on studies and have implemented a temporary policy change to lessen their effect and provide another option for studies.
What’s New: This policy change is currently in effect and provides an option for certain study teams to use Advarra or WCG as a Central IRB (cIRB).
- This change applies to new investigator-initiated studies where the Principal Investigator is from UNMC, Nebraska Medicine, Children’s Hospital & Medical Center, or the University of Nebraska—Omaha.
- This is a temporary change from the policy requiring local IRB review for UNMC investigator-initiated studies.
- This option will be in place until November 1, 2023 unless extended further by the VCR Office/ORA.
- Study teams are still encouraged to use the UNMC IRB, as this change only provides the option to use a Advarra or WCG as the IRB of record.
What to Do: Investigator teams seeking to use one of these commercial IRBs as the IRB of record are required to submit a CIRB application to the UNMC IRB in addition to the application required by the commercial IRB.
Note: Studies should be aware of the following costs associated with using an external IRB: A one-time, $1,500 fee for UNMC review of CIRB studies to ensure compliance with local institutional guidelines. Any additional fees charged by external IRBs for review and use of their services
As a reminder: any human subjects research, whether reviewed by the UNMC IRB or by an external IRB, must adhere to all UNMC HRPP Policies, and must have the approval of all other relevant institutional committees.
For more information, visit the UNMC CIRB website or contact IRBORA@unmc.edu with any questions.
Bruce Gordon, MD Assistant Vice Chancellor for Regulatory Affairs Executive Chair, UNMC IRBs
Russell J. McCulloh, MD Associate Vice Chancellor for Clinical Research Institutional Official, UNMC IRBs
UNMC expanding support for ClinicalTrials.gov registrations
June 30th, 2023
The UNMC Office of the Vice Chancellor for Research and the Office of Regulatory Affairs are updating and expanding support for UNMC’s investigator-initiated studies registered on ClinicalTrials.gov.
To assist UNMC investigators in meeting the complex registration and reporting requirements in ClinicalTrials.gov, the two offices are rolling out an enhanced record monitoring and communication plans that will take effect on July 1.
Key enhancements include:
- Investigators and institutional leadership now will be kept apprised of records with update or reporting problems.
- For any studies experiencing issues with their ClinicalTrials.gov listing, UNMC’s ORA will provide a timeline for correction and assistance as needed.
- A CITI training course is available to UNMC and University of Nebraska at Omaha learners, which provides video instructions for registering, uploading documents and submitting results in ClinicalTrials.gov
- IRB protocols linked to ClinicalTrials.gov records are easily identified via color-coded icons in RSS. Red icons indicate records with problems, and green icons indicate records that currently are compliant.
Over the next few months, additional support and resources will be provided through UNMC’s ORA, including expanded ClinicalTrials.gov information on the UNMC IRB website, a new decision-making tool for determining registration requirements and timing and more robust processes for record management and closure.
The goal is to bring studies registered on ClinicalTrials.gov through UNMC into full compliance with federal regulations, while reducing the complexity and burden of this process on investigators.
The National Institutes of Health and U.S. Food and Drug Administration have increased their enforcement efforts against reports of registration and reporting failures. Penalties can include substantial monetary fines and the suspension of funding for an investigator or the institution.
However, by collaborating with UNMC’s ORA and following the updated process, UNMC and its investigators can achieve full compliance and avoid these consequences.
ClinicalTrials.gov, launched in 2000, serves as a publicly accessible registry for clinical trials. The database provides the public with clinical trial information, while offering researchers access to valuable study data, promoting transparency and informed decision-making in clinical research.
Please email the Office of Regulatory Affairs for more information irbora@unmc.edu.
cIRB webpage update
March 6th, 2023
The cIRB page of the IRB website has recently been reworked. Please feel free to visit the page for updated information as well as a list of helpful forms and links. Keep an eye out for upcoming changes to the sIRB information available on the website!
If you have any questions, please email sirb@unmc.edu or call 402-559-6463.
Reminder regarding Protocol Approval Expiration
January 17, 2023
Investigators and research teams are reminded that annual continuing review is required for research originally approved by the convened IRB (protocols designated FB) as well as most research approved before 2019. As always, reminders will be sent to the PI and the Lead Coordinator and/or Regulatory Contact 60 days and 45 days prior to approval expiration.
If Continuing Review is not submitted and re-approved by the IRB by the expiration date all human subject research activities must stop. This includes new subject accrual, as well as follow-up of existing subjects and data analysis.
If continuation of research activities is in the best MEDICAL interest of already enrolled subjects (that is, the research presents the potential for direct benefit to subjects) you may submit the “Request to Continue Treatment for Enrolled Subjects on Approval Expired Studies" form, available in RSS.
If a Continuing Review application is not submitted within 30 calendar days, the study will be closed, and re-activation of a closed protocol will require submission of a new IRB application.
If all study activities, including all follow-up and data analysis, are completed, and all ClinicalTrials.gov reporting requirements have been met, you must submit a Study Completion Report, available in RSS.
Research which qualifies for Expedited Review (protocols designated EP) and some FB research that is in data analysis or clinical follow-up may not require annual continuing review. The initial approval letter, or a subsequent communication from the Office of Regulatory Affairs, will notify you if CR is not required.
If your research does not require annual CR you must still complete an annual Demographics form. If this form is not submitted within 20 days the protocol will be closed as above.
If you require assistance, please contact us at IRBORA@unmc.edu, or at 402-559-6463
Consent regarding Human Biological Materials
August 26, 2022
Investigators are reminded that obtaining blood samples or other biological specimens for research, whether from a patient or from an employee or other normal volunteer, for use now or in the future, requires IRB approval and written informed consent. This includes drawing blood as “controls” for in vitro assays, unless those assays are performed solely for clinical purposes (and usually in a CLIA certified laboratory).
The IRB is happy to work with you to develop protocols to encompass a number of different activities involving collection of samples from human subjects, and to discuss further. Please contact us at IRBORA@unmc.edu, or at 402-559-6463.
Conducting human subject research without IRB approval and informed consent, including obtaining blood or other biospecimens for any research purpose, is serious non-compliance and may be reported to HHS, FDA, NIH, or other Federal authorities."
cIRB Application Update
June 24, 2022
1 - The CIRB application has been updated to include a contact information sheet which can be automatically generated in RSS. This contact sheet includes the Rights of Research Subjects and Research Questions
- a. Check the box next to personnel you would like to list on the Contact Information sheet. (6/24/22 IRB update 1)
- b. When all personnel have been selected, click “Save.” The page will refresh.
- c. Select “Contact Information Sheet” from the left side menu to generate a pdf that includes a list of authorized personnel and the last 2 pages appended to any consent form. (6/24/22 IRB update 2)
2 - Additional questions have been added to the CIRB application:
- a. “Does this study involve an investigational drug or device?” If yes, prompted for IND/IDE#.
- b. “Will subjects age 18 years of age or younger be included in this research?” If yes, prompted to enter age range.
- c. “Method of Subject Identification and Recruitment” section added for consistency with other IRB applications and to ensure UNMC investigators are following UNMC policies.
- d. “Process of Informed Consent” section added for consistency with other IRB application and to ensure UNMC investigators are following UNMC policies.
Financial Interest Disclosure
June 21, 2022
As of June 21, 2022, PIs and Faculty Advisors must personally complete the Financial Interest Disclosure questions at the time they are signing section I of the application. Though other research personnel may collaborate on completing other sections of the IRB application, no one other than the individuals signing the application will have the ability to answer the financial disclosure questions. This change applies not only to new applications but also to approved applications when a change request is initiated.
Revised Paper Continuing Review Forms
June 17, 2022
The continuing review applications for existing paper applications have been revised. These forms have been revised to remove personnel changes from the continuing review so that the continuing review and personnel changes on paper applications are handled like the electronic applications in RSS. These forms can be found on the IRB website: https://www.unmc.edu/irb/procedures/forms/paper-protocols.html
RSS New Application Update
June 8, 2022
All IRB applications begun after today will no longer require certification and signature by a Resource Reviewer.
Investigators are reminded that they are still responsible for assuring that there are adequate resources available to conduct the research, and to protect the rights and welfare of human subjects.
COVID-19 Update
February 9, 2022
The request to voluntarily defer the start of new protocols and pause face-to-face research activities is now lifted. Investigators should continue to use remote visits, when and where feasible, and review all subjects for potential exposures or risk before any scheduled face-to-face visit. Location or timing of visits may still be impacted by changes in Nebraska Medicine or other clinical facility changes and/or staff shortages.
Reinforcing COVID-19 Guidelines
January 11, 2022
With increasing cases of COVID and COVID breakthrough, we want to clarify/reinforce the new UNMC/Nebraska medicine policies as they are relevant to research spaces. We will make one change in our current policy regarding volunteers in research labs.
For all research programs:
- Masks should be worn everywhere indoors, unless by yourself behind a closed door.
- Cloth masks should not be used anywhere, as per recent Nebraska Medicine and UNMC guidance, unless on top of a surgical/procedural mask
- Conduct any and all research activities remotely, if they can be done remotely, including one on one meetings and seminars
- Visitors should be limited, masked, and escorted with rigorous questions about recent exposures beforehand
- Take turns eating in designated spaces, by yourself or behind a closed door, or socially distanced if in a larger space
- Minimize time unmasked while eating, whether eating alone or in a room where others are sitting
For all lab building and lab-based programs:
- Return to scheduling time on shared equipment and other measures to enhance social distancing in the lab
- Starting Monday Jan 17, research volunteers will no longer be allowed to work in laboratories (such as high school, undergraduate, visiting students) except the following:
- International Scholars who have been already approved through the Office of Global Engagement
- UNMC health professions students or students enrolled in a University of Nebraska course
- High school alliance students in their currently assigned laboratories.
All volunteers must be vaccinated, no exceptions or exemptions, and show evidence of vaccination, per our policy. They should be strongly encouraged to obtain a booster, as soon as they are eligible.
For all face to face clinical research programs:
- Study monitors can come to campus if they are required to, if masked and escorted, and vetted for recent exposures
- Continue to ask research subjects before face to face contact as to recent exposures or symptoms,
Continue to follow your approved biosafety protocol, and all policies of the institution where the research is to be conducted regarding masking, which may now require providing surgical/procedural masks for subjects or accompanying persons.
Demographic Data Requirement Update
January 10, 2022
The Vice Chancellor of Research and the IRB now require that all human research studies provide demographic data (gender, race, and ethnicity) of all enrolled subjects, at time of annual review. This information will be submitted as part of the Continuing Review form, for studies requiring continuing review. For studies not requiring Continuing Review, PIs and Lead Coordinators will receive an email directing him/her to a new Demographics form which has been generated in the FORMS section of your application in RSS.
DocuSign Availability for Electronic Signature
December 6, 2021
DocuSign may now be available for use to document research informed consent in the limited cases where sponsors require its use. This is limited to research NOT subject to the FDA regulations. There may be a charge associated with use of DocuSign; contact Courtney Kennedy in IT for approval or for more information. In addition to DocuSign, investigators may continue to use the RSS e-signature system as previously noted.
Electronic Signature through RSS Expanded Availability
November 11, 2021
Previously only available for studies that were not Federally funded, the RSS e-signature function may now be used for Federally funded research studies also. To request use of this function, please send a message to the IRB via the RSS message portal. Please note, the RSS e-signature function is NOT available for any study that is FDA regulated regardless of funding source.
Remote Consent and Electronic Signatures FAQs
1 - What is remote consent?
- Remote consent refers to the use of techniques like telephone, videoconferencing, or desktop, mobile, or web-based applications (for example, Zoom) as an alternative to face to face discussions in the process of obtaining informed consent.
- HRPP policy 5.3 (Use of a Remote Consent Process)
- To the extent that remote consent facilitates the process of consent, the IRB endorses and encourages its use. However, the Board must approve the specifics of the process.
2 - What is e-Signature?
- e-signature refers to the use of various platforms (like DocuSign, or Adobe Sign) to obtain signatures on a consent form electronically.
- The use of e-signature is independent of the use of a remote consent process. A face-to-face consent process may include an electronic signature, and remote consent may include a “wet” (physical) signature.
- The UNMC IRB must approve the platform used to obtain e-signatures. The specific electronic platforms allowable are dictated by FDA regulations and by Nebraska law.
3 - What is the difference between remote consent and e-signature?
- Remote consent is the process of consent, when consent is not conducted face-to-face.
- E-signature is signing the consent form electronically.
4 - What platforms are allowed for e-signature?
- RSS e-signature is now available for use for Federally funded research. RSS e-signature cannot be used for research which is FDA regulated. In addition, RSS e-signature does not at present support consent forms that require a second signature for optional studies.
- DocuSign, GMO GlobalSign and Solutions Notarius platforms may also be used for Federally funded research, since those platforms are recognized by Nebraska law as equivalent to a “wet” signature. Other platforms may become available in the future, as allowed under Nebraska Law.
- 21 CFR Part 11 (“FDA Part 11”) compliant systems, such as Part 11 compliant DocuSign may be used for FDA regulated research (research involving a drug or device). Note that the standard version of DocuSign, which is not part 11 compliant, is not acceptable.
- REDCap e-signature can only be used for research which is neither Federally funded nor FDA regulated.
5 - How do I access these systems?
- DocuSign, GMO GlobalSign and Solution Notarius: must be licensed by the investigator.
- RSS: the IRB must turn on the e-signature function
- Active Studies: no change request is required. Send a message through the message portal requesting the use of e-signature. Note: If you are adding a remote consent process, a change request will be required.
- For new studies, if you are planning to use a remote consent process, or an e-signature platform (like RSS or DocuSign), the plan must be described in the Consent section in the IRB application.
Changes in the IRB Application
June 10, 2021
Which applications are affected?
Biomedical and Soc/Beh, Human Biologic Material, Medical Records research, Tissue Bank, Data Registry and Humanitarian Use Device protocols.
What are the changes?
- You will be asked to provide an estimate of the time it will take to accrue the target number of subjects for this research.
- You will be asked to describe the timing of consent related to when screening tests are performed.
- The subject identification section has been simplified.
- The process of consent section has been revised.
When do these changes take place?
- These changes went into effect June 9, 2021.
I just started a new application last week and have not yet submitted it, do I need to start another new application?
- No. Much older applications which have not been submitted, however, may need to be updated. If so, an IRB administrator will contact you.
If you have any concerns please contact us by phone 402-559-6463 or email irbora@unmc.edu
IRB Commercial/Industry Fee Changes
March 17, 2021
In line with most other academic medical centers and universities, effective July 1, 2021, the Office of Regulatory Affairs and the IRB will begin charging an annual fee for continuing review of commercial/industry sponsored protocols. This will apply only to protocols submitted after July 1, 2021. In addition, fees for expedited review, and for protocols where UNMC relies on a commercial IRB (like Advarra or WIRB) will increase.
- Full board review of commercial/industry sponsored protocols will remain unchanged at $3000 at time of initial submission. Annual continuing review will now be billed at $1250.
- Expedited review of commercial/industry sponsored protocols will increase from $1000 to $2000 at time of initial submission, and annual continuing review will be billed at $1000.
- Central IRB review (UNMC depending on a commercial IRB) will increase from $1000 to $1500. There is no charge for continuing review for studies using a commercial IRB.
A WBS number must be provided at the time of IRB submission, and billing will occur at the time of IRB review. The protocol will not be released until review fees are paid. Therefore, all investigators involved in commercially sponsored research projects should include the IRB review costs in their grant or contract budget.
If you have any questions regarding this policy, please contact Bruce Gordon, MD
Christopher J. Kratochvil, M.D. Executive Director, Global Center for Health Security Associate Vice Chancellor for Clinical Research, UNMC Vice President for Research, Nebraska Medicine Chief Medical Officer, UNeHealth
Bruce Gordon, MD Assistant Vice-Chancellor for Regulatory Affairs Executive Chairman, Institutional Review Boards Institutional Official, Institutional Biosafety Committee Professor, Pediatrics University of Nebraska Medical Center 987680 Nebraska Medical Center Omaha, NE 68198-7680 bgordon@unmc.edu
Frequently Asked Questions (FAQ)
Below are a number of commonly asked questions. The questions will either provide an answer or will link to the appropriate section of the guidebook. The Page Navigation found in the upper left side of the page can be used to scroll directly to the answer needed and are organized by question. If you do not see your questions listed below, the Book Navigation on the lower left side of the page can be used to browse the various sections. The search bar at the top of the page may also help you navigate to the appropriate section.
Feel free to contact us at irbora@unmc.edu if you cannot find the needed content.
Do I need IRB review?
What application should I use?
What does Exempt mean?
Exempt does not mean exempt from an ethics review, but rather exempt from the federal regulations. Research is exempt from the federal regulations only if it falls under one of the specific exemption categories laid out in 45 CFR 46.
- Information regarding Exempt category research can be found here: HRPP 2.6 (Exempt Research)
- If you would like to know if your project requires IRB review, please use our Determination Survey.
Exempt research protocols - Can I make a Request for Change?
Changes are typically not required for exempt (EX) projects. Changes are only required if an individual being added to the protocol needs access to it in RSS, a new Principal Investigator is being added, or if there is a change to the level of risk that moves the project out of the Exempt category at which point you would need to put through a new application. This is outlined in your approval letter. If more information is needed, please contact your protocol analyst or the IRB by using the RSS message portal for your Exempt protocol.
Is my research project expedited or full board?
The determination of whether a project will be considered Expedited or Full Board will be made by the IRB analyst during the review of the application. If you have a application deadline that requires an expedient review, this can be notated in the IRB application itself in Section 1.4:
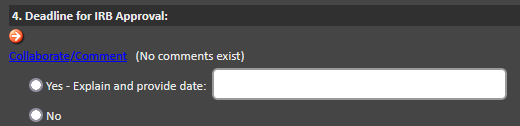
How long does it take for my research project to get approved?
There are many factors involved in IRB review depending on the complexity of the application and how many departments in the Office of Regulatory Affairs must be involved. On average an application will be reviewed between 7 and 28 working days.
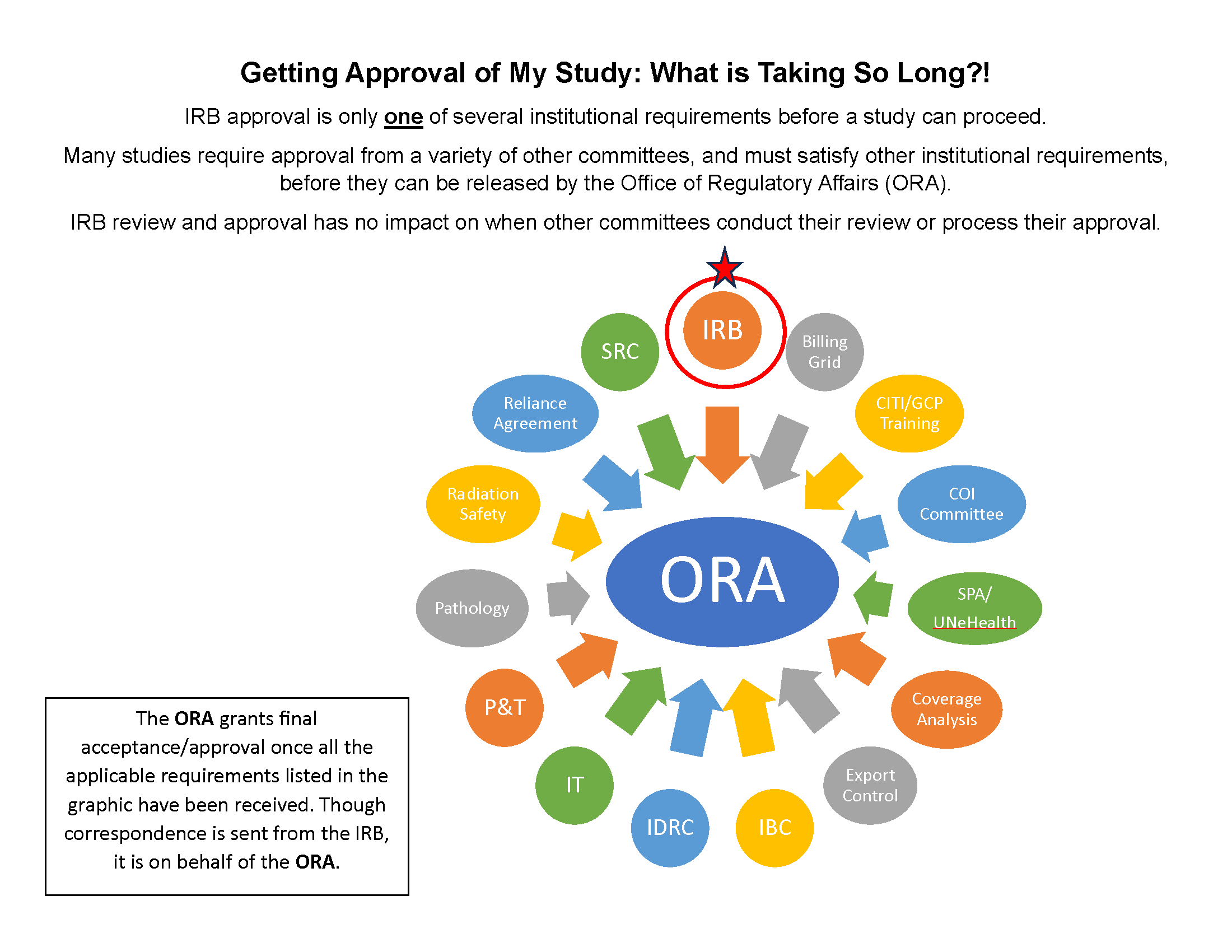
I have a CITI question.
I have an RSS question.
My protocol closes soon, what do I do?
Any protocol which needs to be renewed for another year will fall under one of two scenarios: Continuing Review (Full Board or greater than minimal risk studies) or a Demographic Recruiting Numbers form (EX, EP, CB, and minimal risk). Each will have their own respective form to be completed.
- The Continuing Review period will begin two months prior to the expiration of a study. Notifications will be sent out 60 and 45 days prior to the expiration of the protocol.
- The Demographic Recruiting Numbers form will be generated on the first day of the month a study expires.
- RSS will generate an email notification sent to the PI and Lead Coordinator for both types of renewal.
How do I complete a Continuing Review?
Instructions will be coming soon.
Do I need data from other sites on my Continuing Review (CR)?
- For local studies and cIRB studies - only report UNMC data in the CR.
- For sIRB studies - please contact sirb@unmc.edu
How do I know my Accrual Numbers?
New study applications - when providing information for how many participants are going to be recruited, please provide a solid number value. Avoid using phrases like "approximately" or "around". It is acceptable to provide a larger number than anticipated as investigators can always accrue less participants than applied for, but going above the stated accrual numbers requires submission of a Request for Change and IRB review of stated change. Please take into account that screen failures and withdrawls count as accrual.
Continuing Review/Study Closure/Demographic Recruiting Numbers form - during the yearly renewal (or closure) phase of an investigator's protocol, demographics are often required as part of the process. If demographics are recorded as part of your project, it is advisable to document this information as participants are recruited making this information readily available when reporting is necessary.
How do I close my study?
- For studies requiring a Continuing Review that needs to be closed, a Study Completion Report can be created using the Forms button in RSS.
-
- For studies that require Demographic Recruiting Numbers forms to be closed:
- If the study is in the month of expiration and the form has already been generated, the form can be used to mark the study as Complete.
- If the study is NOT in the month of expiration, send a message to the IRB via the RSS message portal for that protocol. The message will act as official documentation of the request and the IRB will complete the process.
- For cIRB studies: The formal closure of a study is done through the IRB of record. All that needs to be done locally is to send a message through the message portal to the analyst requesting that the application be set to closed. Feel free to upload any pertinent documentation, though not required.


What do I do if I have a problem?
- An investigator can always reach out to the IRB or their study analyst by using the Message Portal in RSS for any particular protocol.
- If the answer to your question cannot be found in the IRB Guidebook, please email irbora@unmc.edu
How often does the IRB meet?
The IRB Boards meet once per month.
- IRB-01 (Adult; first Thursday of every month except January and July)
- IRB-02 (Adult; third Thursday of every month)
- IRB-04 (Pediatric; fourth Tuesday of every month)
- IRB-05 (SIRB; second Friday of every month)
How do I delete a document or consent form?
Once a document or consent form has been deleted, it will be irretrievable. To avoid any accidental deletion of these documents, the request must be performed by a member of the IRB team. Please contact us through the RSS Message portal or at irbora@unmc.edu if this needs to be done.
How do I make a Change Request?
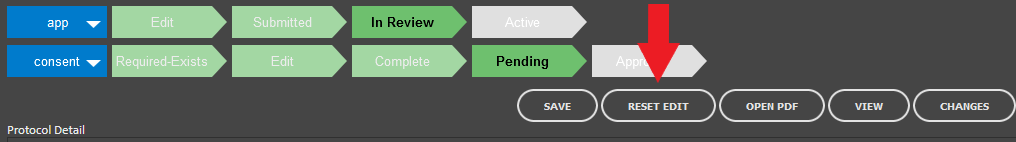
How do I reset my application to edit?
When going into the protocol in RSS, the Reset Edit button should be available at the top of the protocol page. If this does not appear, contact irbora@unmc.edu and the IRB can change the status.
I'm a student, who needs to be listed on my protocol?
The student must be listed as the PI of the project, while the student's faculty advisor must be listed as both Secondary Investigator and Faculty Advisor. The Faculty Advisor role in RSS will appear when question 2.G in the application is answered "Yes".
- If multiple students are involved in a project, only one may be listed as PI. All other students will fill the role of Secondary Investigator. The PI will be the only role capable of signing off on the application and any changes that may occur.
How do I edit a consent form?
I'm leaving UNMC, what do I do with my research before I leave?
- The first option would be to change the Principal Investigator. Often, a Secondary Investigator can be approached to take responsibility for the protocol.
- If no replacement Prinicpal Investigator can be found, the protocol should be closed. Help closing a protocol can be found here.
I have a cIRB/sIRB question.
My project was closed, what do I do?
Please contact irbora@unmc.edu with the protocol number. The circumstances of when and why a protocol was closed will depend on if and how the protocol can be reopened.
How does ClinicalTrials.gov work?
How do I compensate my participants?
I'm trying to add someone to my study in RSS, but I don't see their name.
All UNMC personnel will automatically receive an RSS profile with the creation of their institutional email address. If new employees do not appear in RSS within 48 hours of the creation of their email, please contact irbora@unmc.edu
If personnel do not appear who are members of local institutions (UNO, UNL, and Creighton), when going to https://net.unmc.edu/rss/ have them choose the appropriate institutional portal. This will redirect them to login with their institutional credentials and will sync their access with UNMC's RSS system.
For external institutions, please contact irbora@unmc.edu to determine the best course.
For instructions on how to add or delete personnel in RSS:
What do I need to report with an Incident Report?
How do I know if I need a Data Use Agreement?
When investigators are planning to share or receive data, two things must be considered:
- Is this Research – as determined by the IRB? and
- Does the data include Protected Health Information as defined under HIPAA?
If both of those answers are “yes” it will be necessary to proceed to a DUA.
If it is not research, but rather Quality Improvement (as determined by the IRB) and PHI is being shared, then the Privacy Office will be engaged to determine if a DUA is needed.
If de-identified data is being shared, the IRB does not require a DUA, however one still may be issued if 1) the investigator would like one in place to govern how the data is managed and/or 2) the other institution requires one to be in place.
HRPP (Human Research Policy & Procedures) Manual
The purpose of this policy and procedure is to provide a basic description of UNMC’s Human Research Protection Program (HRPP) through: 1) the Organization’s stated mission, 2) application of ethical principles to guide all human subject research under the oversight of the Organization, and 3) regulatory compliance with all applicable federal, state and local laws.
It is the policy of UNMC that the HRPP will: 1) ensure the rights and welfare of human subjects are protected, 2) evaluate and continually improve the protection of human research subjects, and 3) foster important human subject research in accordance with its mission.
Policies and Procedures Manual (searchable website)
Does my project require IRB review?
To help you determine if your project constitutes Human Subject Research and requires IRB review, you may complete the determination questionnaire by logging into RSS.
The survey can be found in the IRB menu at the top of the page listed under the Application section.
Click the "Am I Doing Research?" button as seen above. This will create a new tab in your browser that will provide the survey page. If the browser tab does not appear, you should see a notification at the top of your browser asking if you would like to allow RSS to generate the survey. All surveys taken by the user will be saved for later access. These can be accessed by clicking on the title of the project where indicated below.
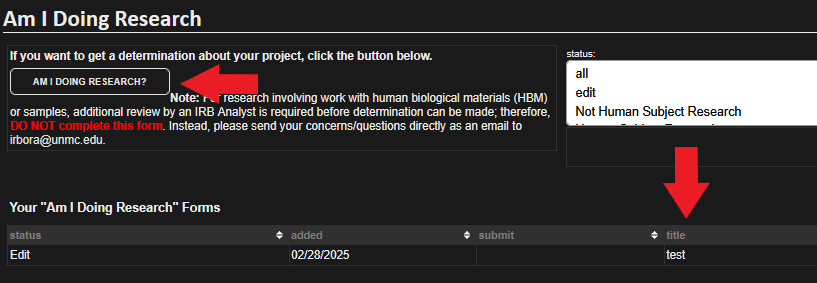
This will allow you to access your past determination surveys at any time in the event that it is needed.
You may also consult the OHRP Decision Flow Charts to help you in making a determination.
Contact our office if you require further information at 402-559-6463 or irbora@unmc.edu.
Which application do I use?
There are eight applications to choose from depending on the type of research that is planned. Please make sure that you select the correct application type for your research. Once started, responses from one application cannot be transferred directly to a different application. Should you have any questions or are unsure as to which option to select, please contact the Office of Regulatory Affairs at 402-559-6463 or irbora@unmc.edu.
Biomedical or Social/Behavioral Research application:
Biomedical Research includes all human subject research performed with intent to develop or contribute to generalizable knowledge (i.e., test a hypothesis and draw conclusions) about human biological systems and processes, including efficacy and safety of preventative, diagnostic or therapeutic methods.
Behavioral and social science research includes all research performed with intent to study behaviors, attitudes and interactions and social processes among and between individuals, groups, and cultures. Generally this category of research has no intent of producing a diagnostic, preventive, or therapeutic benefit to the subject who is not seeking nor expecting a health benefit from the research.
Note: This research will subsequently be divided into therapeutic or non-therapeutic research. Therapeutic research is characterized as research which involves a drug, medical device, technique or other intervention or strategy (including means like diet, cognitive therapy, behavioral therapy, exercise) intended to diagnose, treat or prevent a particular condition or disease.
Human Biological Material (HBM) application:
Research involving the collection of human biological material (HBM) for use for this specific research project, or use of existing HBM (for example, from a biorepository) for a specific research project, or both, with no other type of physical interventions.
Note: HBM studies as described above which would additionally include the collection of medical information via surveys or by chart review should also be submitted using this application. If the sole purpose of this project is to obtain IRB approval for establishment of a tissue bank, complete the Tissue Banking Application.
Tissue Bank application:
Collection of human biological material (HBM) with no other type of physical or laboratory testing for the sole purpose of using the samples for FUTURE research.
Note: If the intent of the protocol is to collect HBM for use for this specific research with storage of left-over material after completion of this specific research, use the Human Biological Material (HBM) application.
Medical Records application:
Research involving the collection of information from medical records whether or not the information already exists or will be recorded in the future.
Note: Medical records research should not be submitted using the Exempt Application.
Data Registry application:
The collection of information/data (typically from medical records) for the sole purpose of using the data for FUTURE research.
Exempt Research application:
This application is only use for research which falls into specifically defined categories under the federal regulations (as outlined below). DO NOT use this application for medical record research.
- Research conducted in established or commonly accepted educational settings that specifically involves normal educational practices.
- Research that only involves educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures, or observation of public behavior (including visual or auditory recording).
- Research involving benign behavioral interventions in conjunction with the collection of information from an adult subject through verbal or written responses.
- Research uses of identifiable private information (either publicly available or subsequently de-identified) that has been collected for some other activity.
- Research and demonstration projects that are conducted or supported by a Federal department and are designed to study, evaluate, improve, or otherwise examine public benefit or service programs.
- Taste and food quality evaluation and consumer acceptance studies.
Note: The Office of Regulatory Affairs (ORA) and the IRB have sole authority to determine whether a research project satisfies requirements for exemption. Contact the IRB Office if you have any questions - [irbora@unmc.edu](mailto:irbora@unmc.edu).
Humanitarian Use Device (HUD) application:
Use of a Humanitarian Use Device (HUD) which is a device that is intended to benefit patients in the treatment and diagnosis of diseases or conditions that affect fewer than 8,000 individuals in the United States per year.
Note: This application should not be used if the HUD is the subject of a clinical investigation in which safety and effectiveness data is being collected to support a pre-marketing approval (PMA) application. The IRB Application for Biomedical Research should be used instead.
Central IRB (cIRB)/Single IRB (sIRB) application:
Multisite research where UNMC will be relying on another IRB for review. This includes commercial IRBs like Advarra and CG-WIRB, Consortium or other Network IRBs, NIH or federal agency IRBs like the NCI CIRB, or other academic institutions. Click here for more information regarding cIRB and sIRB protocols or email sirb@unmc.edu.
Submission deadlines
IRB Submission Deadlines for New Submissions, Previously Tabled Protocols and Requests for Change
- Submission deadlines are for Full Board (FB) protocols only. Review of submissions for Exempt (EX), Expedited (EP), and cIRB (CB) protocols will occur on a first come/first serve basis.
- If an EX, EP, or cIRB protocol is referred to a Full Board, these submission dates will apply. The study team will be notified in the event that this occurs.
- Deadline for Continuing Review submission is the 1st day of the month prior to expiration.
- Full Board Deadlines (printable PDF)
Adult Full Board Meetings
Adult Full Board Submission Deadline – 9 am
| Submission Deadline | Date of Adult Meeting |
|---|---|
| 2/7/2025 | 2/20/2025 |
| 2/21/2025 | 3/6/2025 |
| 3/7/2025 | 3/20/2025 |
| 3/21/2025 | 4/3/2025 |
| 4/4/2025 | 4/17/2025 |
| 4/18/2025 | 5/1/2025 |
| 5/2/2025 | 5/15/2025 |
| 5/23/2025 | 6/5/2025 |
| 6/6/2025 | 6/19/2025 |
| 7/3/2025 | 7/17/2025 |
| 7/25/2025 | 8/7/2025 |
| 8/8/2025 | 8/21/2025 |
| 8/22/2025 | 9/4/2025 |
| 9/5/2025 | 9/18/2025 |
| 9/19/2025 | 10/2/2025 |
| 10/3/2025 | 10/16/2025 |
| 10/24/2025 | 11/6/2025 |
| 11/7/2025 | 11/20/2025 |
| 11/21/2025 | 12/4/2025 |
| 12/5/2025 | 12/18/2025 |
| 12/31/2025 | 1/15/2026 |
PEDs Full Board Meetings
PEDs Full Board Submission Deadline – 9 am
| Submission Deadline | Date of PEDs Meeting |
|---|---|
| 2/13/2025 | 2/25/2025 |
| 3/13/2025 | 3/25/2025 |
| 4/10/2025 | 4/22/2025 |
| 5/15/2025 | 5/27/2025 |
| 6/12/2025 | 6/24/2025 |
| 7/10/2025 | 7/22/2025 |
| 8/14/2025 | 8/26/2025 |
| 9/11/2025 | 9/23/2025 |
| 10/16/2025 | 10/28/2025 |
| 11/13/2025 | 11/25/2025 |
| 12/4/2025 | 12/16/2025 |
| 1/15/2026 | 1/27/2026 |
sIRB Full Board Meetings
sIRB Full Board Submission Deadline – 9 am
| Submission Deadline | Date of sIRB Meeting |
|---|---|
| 2/6/2025 | 2/14/2025 |
| 3/6/2025 | 3/14/2025 |
| 4/3/2025 | 4/11/2025 |
| 5/1/2025 | 5/9/2025 |
| 6/5/2025 | 6/13/2025 |
| 7/3/2025 | 7/11/2025 |
| 7/31/2025 | 8/8/2025 |
| 9/4/2025 | 9/12/2025 |
| 10/2/2025 | 10/10/2025 |
| 11/6/2025 | 11/14/2025 |
| 12/4/2025 | 12/12/2025 |
| 12/31/2025 | 1/9/2026 |
Report a research problem or complaint
Report a problem
Report a problem securely and confidentially through the UNMC Human Subjects Research Comment Portal.
Are you a research subject, staff or investigator who wants to report a complaint or problem to the IRB? By following the comment portal link, you will be directed to a REDCap form where you can enter information about your complaint that will be delivered to the IRB and to the UNMC Chief Compliance Officer. Reports can be made either anonymously or with contact information if you would like to be contacted about your concern.
Alternately, problems can be reported through University of Nebraska EthicsPoint.
By following the EthicsPoint link, you will be directed to a site where you can enter information about your complaint that will be delivered to the UNMC Chief Compliance Officer and University of Nebraska Central Administration.
Research Subjects
The types of complaints/problems that may be reported via this system include:
- Complaints or concerns regarding the conduct (e.g., health or safety concerns) of a research study that you are participating in currently or previously.
- Complaints or concerns regarding the research personnel conducting the study.
Research Staff and Investigators
The types of complaints/problems that may be reported via this system include:
- Complaints or concerns regarding the conduct of other researchers or staff members involved in a particular protocol.
- Complaints or concerns regarding IRB review of protocols or IRB policies and procedures.
IRB Assessment
If you would like to evaluate the IRB or provide feedback on how we have performed, please complete the following form. The form can be sent to the IRB at zip 7830 or sent by e-mail to irbora@unmc.edu.
Procedures
The IRB is committed to making the submission process as smooth as possible. In this section, you will find definitions, explanations and procedures for submitting an application and any other form that may be required during the course of a study.
If you have any questions, please contact us at irbora@unmc.edu
Adverse Event Reporting
Adverse Events (AEs)
An Adverse Event is defined by the NIH as: Any untoward or unfavorable medical occurrence in a human subject, including any abnormal sign (for example, abnormal physical exam or laboratory finding), symptom, or disease, temporally associated with the subject’s participation in the research, whether or not considered related to the subject’s participation in the research.
The IRB requires submission of an AE report form when the event is unexpected and related/possibly related to the research. Adverse events occurring on a study which satisfy these criteria must be submitted to the IRB within the timeline specified in the policy. Any death, which occurs while the subject is being treated on protocol or occurs within 30 days of completing research related interventions, must be reported immediately if it meets the reporting criteria.
For more information, please refer to HRPP policy 8.1
AEs will be submitted through the RSS system.
External Adverse Events
External Adverse Events are defined as: adverse events that occur at a site under external IRB oversight. External AEs are not reported to the IRB unless they require a change in protocol or revision of the consent document. These are not reported on an Adverse Event report form. The external AE report (i.e., IND Safety Report) is used as justification for the required changes.
External Adverse Device Effects (UADEs)
Unanticipated Adverse Device Effects (UADEs) are defined as: Unanticipated adverse device effect means any serious adverse effect on health or safety or any life-threatening problem or death caused by, or associated with, a device, if that effect, problem, or death was not previously identified in nature, severity, or degree of incidence in the investigational plan or application (including a supplementary plan or application), or any other unanticipated serious problem associated with a device that relates to the rights, safety, or welfare of subjects.
External AEs for device studies must be reported to the IRB (in no case no later than 5 business days following PI notification from the sponsor that the event occurred) in accordance the requirements of 21 CFR 812.150(b)(1).
The PI should submit the report received from the sponsor along with any required Request for Change.
Once the status of a study is changed to “completed,” the IRB will no longer accept external UADE reports except under circumstances where the report involves important new risk information.
For more information, please refer to HRPP policy 8.1
For any questions, please email irbora@unmc.edu
ClinicalTrials.gov (CT.gov)


Check back to this page for more updates regarding clinicaltrials.gov information.
Please contact oract.gov@unmc.edu for more information.
When requesting a new user account for ClinicalTrials.gov, please provide the following information:
- Preferred user name
- Institutional email address or, if none, other email address
- Office phone number
Once the account is created, ClinicalTrials.gov will send an email with login information.
Note: For Student Principal Investigators, if your research study will be registered on ClinicalTrials.gov, please list your Faculty Advisor as the Responsible Party and yourself as Record Owner.
CITI Training Course for ClinicalTrials.gov
A new CITI training course is available to UNMC and UNO learners that provides video instructions for registering, uploading documents, and submitting results in ClinicalTrials.gov. Currently optional, the course is highly recommended as a guide to investigators new to ClinicalTrials.gov requirements and to experienced investigators needing a refresher.
- Login into your UNMC or UNO CITI account
- Scroll down to 'Add a Course'
- Click the box for Protocol Registration and Results Summary Disclosure in ClinicalTrials.gov
- Click 'Next'.
When you have successfully completed the course, please email a copy of the Completion Certificate to oract.gov@unmc.edu.
ClinicalTrials.gov Icons in RSS
IRB protocols that are investigator-initiated and registered on ClinicalTrials.gov with an NCT# will now be denoted by an icon in RSS.
A green icon means no problems are currently identified by ClinicalTrials.gov on the record associated with the study.

A red icon means ClinicalTrials.gov has identified problems on the associated record.

If your study has a red icon, please login at https://register.clinicaltrials.gov to correct the problem(s). Icons are updated daily, Monday-Friday, so once problems are resolved, the icon will be green after the next daily update. If you have difficulty correcting a problem in https://register.clinicaltrials.gov, please contact oract.gov@unmc.edu for assistance.
Outstanding problems with the ClinicalTrials.gov record may delay the review and approval of IRB submissions. Please ensure all problem records are addressed as soon as possible (UNMC HRPP Policy 1.29).
Process for updating a record
Whenever a ClinicalTrials.gov record is updated, the process must be completed by approving and releasing the update.
Steps for completing an update of any type are displayed in the “Record Status” at the top of the record.

The “Next Step” box is displayed immediately below which describes the next action needed.
Any problems with the update are listed in the “Next Step” box and may include:
- Correct Error(s)
- Enter Results
- Finish Protocol/Documents/Results section
- Address Review Comments
Once the update problems are resolved, the next step is to click the “Entry Complete” button:

The following step is to review the update, then click the “Approve” button:

The last step is to click the “Release” button if you are the Responsible Party for the record:

If you are the Record Owner, this “Next Step” box will be displayed, and the Responsible Party will need to login and release the update.

When any problems with the update are resolved and the update has passed PRS review, the update is released to the public ClinicalTrials.gov site.

All the steps on the “Record Status” will be highlighted in blue.
Posting Consent Forms
For any clinical trial conducted or supported by a federal agency or department or agency, Federal Regulations require the awardee of a grant to post one IRB approved informed consent form used to enroll subjects on a publicly available Federal Web site.
“Clinical trial” means a research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of the interventions on biomedical or behavioral health-related outcomes.
The PI must post:
- Only if the organization (UNMC, Nebraska Medicine, UNO or CHMC) is the lead site or grantee organization.
- Only clinical trials (defined above). As a general rule, if you reported to clinicaltrials.gov then you will also have to post the consent form.
- Only if conducted or supported by a Federal department or agency.
The PI only needs to post ONE consent form used to enroll subjects anytime during the course of the study.
The consent form must be posted no later than 30 days after the last subject is enrolled.
If your study is utilizes the Clinical Trials Monitoring System (CTMS) you will receive notification when your last subject is enrolled. The notification includes instructions about the requirement, and how to post to clinicaltrials.gov.
If your study does not utilize CTMS it is your responsibility to track subject enrollment, and post no later than 30 days after the last subject is enrolled.
Specific instructions on how to register with ClinicalTrials.gov and upload documents can be found here.
Emergency Treatment
The contact list for Emergency Treatment authorization can be found here in RSS.
Under certain circumstances, a physician may treat a patient with an investigational (non-FDA approved) drug, biologic or device, or treat a patient utilizing a non-IRB approved protocol; Pursuant to FDA regulations, the patient must be suffering from a serious, life-threatening or debilitating illness for which there is no satisfactory treatment alternative(s) and there must not be sufficient time to obtain full IRB review and approval. Emergency treatment as defined here is not research. The FDA regulations do not provide for expedited IRB approval in emergency situations.
UNMC/Nebraska Medicine policy requires the IRB be notified prior to such use, by contacting the IRB office. This notification is not IRB approval. The IRB will only state it is aware of the proposed use and considers the use to meet the requirements of 21 CFR 56.102(d), 21 CFR 56.104(c), and the criteria in HRPP 6.4 Emergency Use of a Test Article, section 5.0 of the full policy.
The investigator is still required to obtain informed consent of the patient or the patient’s legally authorized representative. The consent form must contain appropriate elements structured to reflect that consent is for treatment purposes as opposed to research. View a sample Consent Form for Emergency Treatment
A useful guide can be found here: Emergency Use vs Expanded Access
The UNMC Emergency Use of a Test Article Report form can be found in RSS and a signed copy of the consent form must be submitted to the IRB within 5 business days following the treatment.
Exempt Studies
It is understood this project will be conducted in full accordance with all applicable HRPP Policies. It is also understood that the ORA will be immediately notified of any proposed changes for your research project that:
- Affect the risk-benefit relationship of the research
- Pose new risks which are greater than minimal
- Constitute a new risk to privacy or confidentiality
- Involve sensitive topics (including but not limited to personal aspects of the subjects behavior, life experiences or attitudes)
- Involve deception
- Target a vulnerable population
- Include prisoners or children
- Otherwise suggest loss of the exempt status of the research.
No changes to exempt studies are required to be submitted for review, which is outlined in your approval letter. If you feel a change is warranted, please contact our office utilizing the message portal of the application in question.
You are encouraged to contact the ORA to discuss whether changes to exempt research requires review by the IRB or please view HRPP policy 2.6 (Exempt Research).
Incidents (Non-compliance/Problems)
Per HRPP policy 8.4 non-compliance (NC) involving the PI and/or study personnel must be promptly reported to the IRB. This non-compliance could involve failure to comply with the Federal Regulations related to the protection of human subjects of research, HRPP policies, or the requirements/determinations of the IRB.
NC and problems are reported to the IRB by submission of an Incident Report Form. The Incident Report form is located in the Forms section of RSS. The PI is responsible for ensuring that the required reports are submitted promptly following discovery of the incident in accordance with HRPP policy 8.4.
If you have any questions regarding this submission process, please contact the IRB Office at irbora@unmc.edu
Protocol Deviations
A Single Subject Protocol deviation is a change in an IRB-approved protocol which is permitted for an individual subject when it is in the best interest of that subject and/or is necessary for research purposes (e.g., data completion). Protocol deviations are classified as either “minor” or “more than minor.” Once the form is received, the deviation will be reviewed and processed by the IRB. Deviations may be approved in one of two ways:
- Deviations that are minor are eligible for expedited review under the provisions of HHS regulations at 45 CFR 46.110(b)(2) and FDA regulations at 21 CFR 56.110(b)(2), as applicable.
- Deviations that are more than minor do not qualify for expedited review and therefore must be reviewed by the full IRB.
Once the deviation is approved, an approval letter for the Single Subject Protocol Deviation will be attached to the electronic IRB application. The PI and Lead Coordinator will be notified via email when this occurs.
To obtain Single Subject Protocol Deviation approval, a Single Subject Protocol Deviation Request must be submitted PRIOR to the implementation of the deviation. The instructions for submission are below:
RSS Studies & Existing Paper Format Studies
The Single Subject Deviation form can be created by pulling up the protocol in RSS. The 'Forms' section will be found in the left-side menu. When clicking on 'Forms' a list will appear allowing you to choose the 'single subject protocol deviation request'. Please follow the instructions provided to complete the form and once competed the PI will electronically sign and click 'SUBMIT'.
If you have any questions regarding this submission process, please contact the IRB Office at irbora@unmc.edu.
Submission Process
Full Board Review (initial submission)
Investigators will be notified of the assigned IRB# by email. Applications for Full Board Review will be reviewed at the next possible IRB meeting. Submission Deadlines can be found here.
These documents must also be attached in the "ADD DOCUMENTS" section of the online application at the time of electronic submission:
New Submissions (Initial)
- Subject Recruitment Material (as applicable)
- External study site approval letters (as applicable)
- Full protocol (as applicable)
- Investigator's Brochure (as applicable)
- Grant application (as applicable)
- Clinical Trial Master Matrix (as applicable)
- Other relevant material (e.g. surveys) (as applicable)
Tabled — Re-Submissions
- Investigator's response letter
- Other revised materials (as applicable)
Expedited Review (initial submission)
Certain studies involving no more than minimal risk may qualify for expedited review status under 45 CFR 46.110 or 21 CFR 56.110. View a list of categories which may qualify for expedited review.
Expedited review of new protocols are handled through the electronic submission only. New submissions eligible for expedited review will be reviewed by the IRB Analyst with appropriate confirmation by the Executive Chair/designee and the investigator will be informed of the IRB’s decision by email.
Exempt Protocols (initial submission)
Research activities in which the only involvement of human subjects will be in one or more of the categories specified by 45 CFR 46.101(b) are exempt from the requirements of 45 CFR 46. The exempt categories do not, however, apply to research involving deception of subjects, sensitive behavioral research, or to research involving pregnant women, prisoners, individuals who are decisionally impaired and other subject populations determined to be vulnerable.
Reviews of new Exempt protocols are handled through the electronic submission only. New Exempt submissions will be reviewed by a member of the Office of Regulatory Affairs (ORA) staff and the investigator will be informed of the ORA's decision by email.
Request for Change
Any proposed change in a research activity must be reviewed and approved by the IRB prior to implementation except when: 1) a change is necessary to eliminate an apparent immediate hazard to the subject(s), or 2) a subject needs to be advised immediately of significant new information. Administrative changes do not require IRB review and can, accordingly, be approved by ORA.
For studies submitted in electronic system, follow these steps:
- Reset the application to Edit
- Make changes to the IRB application and click SAVE
- Revise consent documents (if applicable) and click COMPLETE
- Upload any applicable documents using the Add Document function
- Click CHANGE REQUEST on the left-side menu and complete
- Sign the application (PI)
- Click SUBMIT
For Studies NOT Submitted in Electronic System, follow these steps:
- Complete, sign (PI), and upload the request for change form found here.
- Revise the current approved application using track changes
- Sign Section I of the application (PI)
- Revise current approved Consent document as applicable using tracked changes
- Complete and sign the appropriate Request for Change
- Upload all documents associated with this change (e.g., application, consents, Request for Change, etc) using the Add Document function
If you have any questions regarding this submission process, please contact the IRB Office.
Continuing Review Overview
Federal regulations require certain types of research undergo continuing review at least annually. Review and approval of continuing review must occur before the expiration date listed on the initial approval letter and, subsequent continuing review approval letters as the “valid until” date. Courtesy email reminder notifications are sent approximately two months before expiration and again two weeks later.
Federal regulations prohibit the IRB from granting extensions or temporary approval beyond the expiration date. Should expiration of IRB approval occur, all study related activities must cease as of the date of expiration.
A “Request to Continue Treatment for Enrolled Subjects on Approval Expired Studies” form must be submitted and approved to allow currently enrolled subjects to continue to receive study treatment. This form is available through the “Forms” link on study pages in RSS.
CR Electronic Submission
- Complete, sign (PI), and submit the continuing review form within RSS (located on the left side menu under “Forms”)
- Upload the following documents, as applicable, using the “Add Document” function:
- Last signed consent form (only if a subject was consented or reconsented since the last continuing review). These are no longer to be redacted (i.e. subject identifiers do not need to be blacked out).
- Scientific Review Committee (SRC) CR approval letters
- DSMB or other safety review reports
- Progress reports
- Publications
CR Existing Paper Protocol Submissions
- Complete, sign (PI), and upload the continuing review form found here.
- Upload the following documents, as applicable, using the “Add Document” function:
- Consent documents (Word format and clean to be date stamped)
- Last signed consent form (only if a subject was consented or reconsented since the last continuing review). These are no longer to be redacted (i.e. subject identifiers do not need to be blacked out).
- Scientific Review Committee (SRC) CR approval letters
- DSMB or other safety review reports
- Progress reports
- Publications
Submission Deadlines
Studies requiring Full Board (convened IRB meeting) review, continuing review applications should be submitted 4-6 weeks before expiration.
Studies requiring expedited review, continuing review applications should be submitted four weeks prior to expiration, to allow time to address any required modifications.
Studies that do not require continuing review, are required to submit an annual update, which includes a report of subject demographics. Emails requesting the annual update are sent out the month the study will expire.
If you have any questions regarding this submission process, please contact the IRB Office at irbora@unmc.edu.
Education & Resources
The goal of the IRB Education Program is to facilitate research involving human subjects from initial submission to study completion through didactic and practical education. Whether it is for investigators, research study personnel, students or other institutional representatives, information is explained in a manner that fits the audience. By using a variety of delivery methods, such as lectures, webinars, live-streams, bulletins, one-on-one meetings and department in-service, from new student to seasoned investigator, our objective is to offer education of:
- The history and regulation of research ethics
- The local submission requirements and process
- The common pitfalls to improve the efficiency of the submission process.
The IRB is here to help!
All educational options will be tailored to meet the specific needs of the target audience, whether it be one-on-one about a specific research protocol or a lecture to a class regarding a general overview of research ethics and the IRB process.
If you would like to schedule a one-on-one meeting, department in-service, class lecture, Q&A session or any other type of IRB education, please contact IRB staff for assistance at irbora@unmc.edu
HRPP Investigator Guidance Series
This page serves as a hub for all of the Investigator Guidance Series documents. Each document is an abbreviated version of one of our HRPP Policies and Procedures intended for investigators, coordinators, and other study team members. This page is a good starting point for any study team member with a question about a policy on a specific topic. A link to the full policy/procedure is included in each document.
Investigator Resources
Regulations:
- Common Rule (45 CFR 46)
- eCFR: 21 CFR Part 50-Protection of Human Subjects
- eCFR: 21 CFR Part 56-Institutional Review Boards
National Institutes of Health:
- NIH Home Page
- Office of Recombinant DNA Activities (ORDA)
- Office of Grants and Contracts
- National Human Genome Research Institute
UNMC Links:
- General Counsel's Memo on Mandatory Reporting of Child Abuse and Related Statute of Limitations
- Institutional Biosafety Committee (IBC)
- Animal Care and Use Program (IACUC)
- Sponsored Programs Administration (SPA)
Food and Drug Administration:
- FDA Web Site
- FDA - Center for Drug Evaluation and Research (CDER)
- FDA - Center for Devices and Radiological Health
International Standards:
Other Federal Agencies:
- National Archive and Records Administration
- Office for Civil Rights
- Department of Health and Human Services
Organizations and Other Items of Interest:
- Public Responsibility in Medicine & Research (PRIM&R)
- National Bioethics Advisory Commission (NBAC)
- American Society for Bioethics and Humanities
IRB History and Principles:
- “What Makes Clinical Research Ethical?” Emanuel, et al; JAMA 283(20):2701, 2000
- THE BELMONT REPORT: Ethical Principles and Guidelines for the Protection of Human Subjects of Research(UNMC)
- The Belmont Report(HHS)
IRB Conference Content
2024
- 2024 IRB Conference Introduction (Bruce Gordon)
- 2024 IRB Conference Conclusion (Bruce Gordon)
- Big Data and Ethics (Scott Campbell)
- Considerations for Patient Safety in Studies of Psychedelics and other Non-Ordinary States of Consciousness. (Lou Lukas)
- De-Centralized Trials (Megan Singleton)
- Let's Review Some Research (Nancy Olson/Nichelle Cobb)
- Research on the Edge (Bruce Gordon)
2023
- 2023 IRB Conference Introduction (Bruce Gordon/Russell McCulloh)
- Participant Compensation (Joe Brown/Dustin Krutsinger)
- Personal Narrative and Research Ethics (Gigi McMillan)
- Re-examining the IRB's Role in Protecting Research Subjects (David Strauss)
- Uncovering Bias in Artificial Intelligence (John Windle)
- Understanding CBPR (Keyonna King/Russell McCulloh)
2022
- 2022 IRB Conference Introduction (Bruce Gordon)
- Genetics, Research, and Diversity (Omar Rehman/Kristi DeHaai)
- Informed Consent & Teach Back Workshop (Elizabeth Bankert)
- Intro to the IRB (Bruce Gordon)
- Readability is Fundamental (Nichelle Cobb)
- Review of Research Involving Products with EUA (Kindra Cooper)
- Risk of Harm to Non-Subjects (David Borasky)
Mental health considerations
MENTAL HEALTH CONSIDERATIONS FOR RESEARCHERS
DECEMBER 2023
EXPLANATION OF RISKS:
- Be clear in application and ICD about risks associated with mental health assessments (cognitive status assessments, IQ screens, mental health assessments, exploitation/abuse/violence assessments, and drug testing).
- Describe how and by who mental health assessments and outcomes are reviewed and reported.
- Remember to report psychiatric adverse events, including serious adverse events, appropriately.
SELF-REPORT MEASURES:
- Protocols using subject self-reports that ask about depression, worthlessness/guilt, and quality of life, should include a process of review by personnel with plan to notify investigator of pertinent positives.
- Protocols using subject self-report reports with items specifically addressing self-harm or suicidal ideation, or related items indicating a subject may be at risk, should have a mechanism for responses to be reviewed in REAL TIME so action can be taken as appropriate.
- Protocols using remote self-reports (Ipad, EMA device, web-based, etc.) should include a mechanism for notification of the investigator or designated member of the study team when threshold responses are received so that REAL TIME management can occur.
INVESTIGATOR-ADMINISTERED MEASURES:
- Investigator-administered measures of psychiatric symptoms should be completed by those with appropriate training.
- If the study team does not have the specific expertise, consider consultation with psychiatry or psychology colleagues.
PHQ-9:
- PHQ-9: Suggestion to align with Suicide Risk BPA’s used by NM PCMH clinics rooming staff starting 8/8/2022:
- if >14 and + response to question 9 = refer for emergency eval
- if >14 and – response to question 9 = refer for mental health consult
- if <14 and + response to question 9 = further assessment needed; refer as appropriate
- if <14 and – response to question 9 = no further specific intervention
The Columbia Suicide Severity Rating Scale-Revised (CSSRS-R):
- Baseline (“lifetime”) and “since last visit” versions available on-line.
- Validated and available in Spanish.
- Use of this scale should include training for non-mental health providers as it explores suicidality in a very thorough manner:
- To complete the C-SSRS Training for Clinical Practice, visit http://c-ssrs.trainingcampus.net/
- General information, go to http://cssrs.columbia.edu/
RESOURCES:
- CURRENT – include 988 for the suicide hotline, don’t give numbers to agencies now closed (911 is still ok to use).
- ACCURATE—know the policy for referral to the Department of Psychiatry, procedures for accessing ER, the Psychiatric Emergency Service (PES). Consider age- and/or diagnosis-appropriate services (e.g. Nebraska Family Help Line [1-888-866-8660]; Professional Partners-Region specific).
- LOCAL—while resources are limited in some areas of the state, please make sure you list the ones close to the subject’s home.
UNMC/NE MEDICINE PSYCHIATRY SERVICES:
- Psychiatry (ADULT) accepts referrals from PCP’s within the system.
- C/A psychiatry not limited to UNMC/NE Med providers.
- Behavioral Health Connections team (402-552-6007) facilitates referrals to community agencies.
- When referring to the “PES” (Psychiatric Emergency Service), understand that patients still must go through the regular NE Med ER or Bellevue Medical Center ER first.
PSYCHIATRY SERVICES FOR CHILDREN
- Immanuel (CHI) ER is primary location for inpatient triage for children/teens; other ER’s may transfer there if hospitalization is needed.
- Bryan LGH (Lincoln) has inpatient care for children/teens as well as emergency shelter placement.
- Boys Town (Grand Island) has emergency shelter placement.
- Mercy (Council Bluffs) will accept NE youth (even Medicaid if no NE beds available).
- Boys Town has an inpatient unit—triage through Methodist ER’s.
PSYCHIATRY SERVICES FOR STUDENTS:
- For UNMC students: call UNO Health Center, 402-554-2374 (select option 2 to leave message for the nurse for scheduling).
- For UNO students: Call CAPS 402-559-7276 (initial appointments are covered by student fees).
- Gender and Sexuality Resource Center (GSRC): Confidential and free, Student Life Center 2031. Call 402-559-7276.
KEARNEY COMMUNITY RESOURCES
- S.A.F.E. Center: 24/7 hotline 1-877-237-2513
LINCOLN COMMUNITY RESOURCES
- Voices of Hope: Crisis hotline 402-475-7273 (non-emergencies, 402-476-2110)
NORFOLK COMMUNITY RESOURCES
- Bright Horizons: call 877-379-3798 or text 402-370-8817
SCOTTSBLUFF COMMUNITY RESOURCES
- Doves Program: call 308-436-4357 or 866-953-6837; text 515-599-6620
NATIONAL RESOURCES
- National Domestic Violence Hotline: 1-800-799-7233, TTY 1-800-787-3224
- National Suicide Prevention Lifeline: Text or Call 988
- Trans Lifeline: 1-877-565-8860
Miscellaneous Resources
Below investigators will find a variety of resources that may assist with research goals:
- Allowable Costs Related to Participant Inclusion Activities
- Emergency Preparedness / Continuity of Operations Plan (EP/COOP)
Training
The topics found below will provide various learning resources for common IRB items.
If there is a topic that you would like to learn more about but do not see it listed below, please contact us at 402-559-6463 or at irbora@unmc.edu.
CITI Training
![]()
What is CITI?
Collaborative Institutional Training Initiative (CITI) certification is an institutional requirement for all personnel engaging in Human Subject Research (HSR). It includes three primary modules that are required by the institution to participate in conducting various types of research: Group 1: Biomedical Research, Group 2: Good Clinical Practice (GCP), and Group 3: Social & Behavioral Research.
Faculty, employees, students and other institutional representatives at UNMC, Nebraska Medicine, CHMC, and UNO are required to complete the Human Subjects Research (HSR) course via CITI if they will be working on a research project that involves human subjects. It takes approximately 1-2 hours to complete a Basic course. The training does not have to be completed in one sitting, but can be spread out over time if needed.
Instructions
A PDF with instructions for how to navigate CITI can be found here.
Basic or Refresher?
The Basic course is designed to establish certification and should be taken when:
- No previous CITI training has been completed, or:
- Prior CITI certification has been expired for a period greater than three years
The Refresher course is designed to re-establish certification for three years and should be taken when:
- The Basic course of a particular group has already been taken, and:
- Recertification is required, but has not been expired for a period of three or more years
Course required based on type of research:
Group 1: Biomedical Research – Investigators conducting research about human biological systems and processes, including efficacy and safety of preventative, diagnostic or therapeutic methods must take this course. Types of research:
- Clinical trial using a drug, medical device, technique or other intervention or strategy (including non-physical means, like diet, cognitive therapy, etc.) to diagnose, treat or otherwise study a particular condition or disease.
- Non-clinical biomedical research to study normal or abnormal physical or physiologic processes (for example: gait and balance testing, biomechanical assessments, etc.).
- Research involving medical records or data registries.
- Research involving human biologic materials.
Group 2: Good Clinical Practice (GCP) – Investigators conducting clinical trials funded by NIH, or utilizing an FDA regulated drug, device, or biologic must take this course. A clinical trial is defined as “a research study in which one or more human participants are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of those interventions on health-related biomedical or behavioral outcomes”.
Investigators conducting these types of trials must also take the Biomedical course (Group 1).
This GCP course meets the minimum criteria for training identified by some sponsors. (Check to see if your sponsor is listed.) If so, your CITI completion report can be supplied to the sponsor to meet their GCP training requirement.
- Behaviors, attitudes, and interactions/social processes among and between individuals, groups, and cultures.
- Generally, this category of research has no intent of producing a diagnostic, preventative, or therapeutic benefit to the subject who is not seeking nor expecting a health benefit from the research.
- This course is primarily taken by students at UNO, although it is common for Nursing Program students to be required to take this as well.
Researchers/Students transferring from other institutions:
Please email all completion reports for previously completed CITI training courses to irbora@unmc.edu. CITI courses are unique from institution to institution and transcript comparison will be required. Only the completion report shows the modules required for transcript comparison. Once previous training has been updated, the IRB will determine if any additional training or Refresher courses will be required.
It is highly recommended that you email the Completion Report prior to beginning the Refresher course. Please do not send completion certificates.
Collaborators with UNMC:
Any independent entity collaborating with UNMC for the purposes of research must also complete CITI training as required by the institution. When registering for CITI, please affiliate with UNMC/UNO and do not register as an independent learner.
If you need assistance with your username and/or password from an institution other than UNMC/UNO, you can contact:
CITI Support: 888-529-5929 (9:00 a.m. to 7:00 p.m. EST/Monday – Friday).
If you have more than one CITI account, you can request that they be merged by calling CITI Support. Once merged, all training completed will be available under one account.
For all questions regarding CITI training, please contact the IRB Office at 402-559-6463 or email irbora@unmc.edu.
Community Partners
All individuals that work on human subject research projects must complete human subject research training. Faculty, students, or employees of UNMC, NM, CHMC, UNO, BMC, or another academic partner institution are required to take CITI training. Community partners who collaborate on research projects may complete the CIRTification program as an alternative to the CITI Program.
Who are Community Partners?
Community partners are non-academic personnel (e.g. community leaders, representatives from supporting community organizations) engaged in research requiring IRB approval. This program is specifically and only for community members collaborating on human subject research studies.
Who is NOT considered a Community Partner?
Students or staff affiliated with UNMC, NM, CHMC, UNO, BMC, or research staff affiliated with other private entities directly engaging in human subject research.
What is CIRTification?
CIRTification is a training program in human research protections created by the University of Illinois Chicago CCTS that is tailored to the unique roles of community research partners. It is interactive and relevant to the roles and responsibilities that community partners have in research projects. The program considers community partners’ limited experience with research, discusses key concepts in research ethics and responsible conduct of research in plain language, and focuses on applying knowledge to real-life scenarios. Ideally, CIRTification Online will not only teach community partners about the importance of protecting research participants, it will also empower them to be active contributors to their respective research teams.
CIRTification introduces learners to the basics of the research - the terminology, people, and methods. It reviews the history of research abuses that have informed current ethical principles, rules, and regulations.
The training program also covers standard and best practices for:
- Recruitment and informed consent
- Collecting and protecting data
- Handling changes that may arise during participant interactions
- Reviews the role of the Institutional Review Board in protecting the right and safety of human research participants
What can I expect?
The program takes about 3-4 hours total to complete and can be completed in multiple sessions. The course is currently available in English, Spanish, and Haitian Creole. The course includes audio, video, text, and interactive activities.
Learners will complete a knowledge quiz at the end of the program and receive a date-stamped certificate of completion. Please save a copy of this certificate for your records and email a copy to IRBORA@unmc.edu.
Online Training Instructions:
Please follow the instructions below to enroll in CIRTification. Please select University of Nebraska Medical Center only from the list.
- Go to https://training.ccts.uic.edu/
- Click “Register” in the top right-hand corner.
- Select “I am not from UIC”.
- Complete the registration form. Under “Site”, select “University of Nebraska Medical Center”.
- Once the form is filled in, click “Register” at the bottom of the form.
- Visit the Course Catalog. Information about CIRTification will appear. Click “Learn More”.
- Click “Enroll” to start the CIRTification course.
Frequently Asked Questions (FAQ):
| Who can I contact for help? |
|---|
| For assistance, place contact: Megan Berger at mberger@unmc.edu |
| How long is this training valid? |
|---|
| The training is valid for three years. |
| How do I access my completion certificate? |
|---|
| After completing the quiz at the end of the training, the option to print and save a date-stamped certificate of completion is available. If for some reason you missed this or are unable to complete this step, Megan Berger can access the completed training certificate. Email her at mberger@unmc.edu. |
CIRTification is funded through the University of Illinois at Chicago Center for Clinical and Translational Science and supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1TR002003. We encourage mention and citation of CIRTification Online in grant proposals, conference presentations, published manuscripts, and other reporting. Suggested citation: CIRTification Online: Community Involvement in Research Training. University of Illinois at Chicago, Center for Clinical and Translational Science.
Consent Forms
Capacity to consent
Consent Form Readability
Recent changes to the Federal Regulations governing human subject research (the “Common Rule”) have included a focus on improving the readability of consent forms and include regulations requiring understandable language, and organization and presentation of information that facilitates understanding.
In response to these requirements, beginning October 31, 2019, consent forms must satisfy minimum readability standards. We are working on a way to receive proof of PRISM training. Therefore, until further notice, we are not requiring physical proof, but written assurance.
Though we expect to extend the standard to other sections, initially only the readability of the Invitation and Summary section will be assessed.
This section must have Flesch Kincaid reading level ≤8 and Flesch Reading Ease ≥60. Readability may be scored within the RSS application by clicking on the “Readability” button.
CFs not meeting these minimum readability measures will be returned to the investigator for modification.
To assist investigators and their staff in developing necessary skills to write effective consent forms, the PI and the person responsible for writing the consent form, must complete online training through PRISM (Program for Readability in Science and Medicine). The hour-long training covers health literacy and readability, plain language strategies and examples, and interactive editing examples and exercises.
For more information on process, readability standards, and readability tips, see below.
Readability Assessment Process:
-
Readability can be assessed within the RSS application by clicking on the “Readability” button. This will open a new page with assessments of Flesch-Kincaid reading level and Flesch Reading Ease for the invitation and summary section.
-
In computing reading levels, technical terms (especially drug names and names of medical procedures) may unavoidably adversely affect the score. You may choose to use the name once, and then say "the test drug" or "the operation" subsequently. However, in all cases, an effort should be made to use a simpler word or phrase.
-
The IRB recognizes that sections of some consent forms may be of such a technical nature that it may not be possible to keep to an 8th grade reading level. In these situations, the invitation and summary at least must meet the standard described above, and the investigator must describe what additional tactics will be used to assure and assess comprehension.
Readability Standards:
- The Flesch–Kincaid readability tests are readability tests designed to indicate how difficult a passage in English is to understand. There are two tests, the Flesch Reading Ease, and the Flesch–Kincaid Grade Level. Although they use the same core measures (word length and sentence length), they have different weighting factors.
- The results of the two tests correlate inversely: a text with a comparatively high score on the Reading Ease test should have a lower score on the Grade-Level test.
- Many other measures of readability exist. SMOG (Simple Measure Of Gobbledygook) is a more exacting measure of readability, and accurately scores for the grade level required for complete text comprehension. (The Flesch-Kincaid formula grades for less than complete comprehension). SMOG demonstrates strong correlation with the required reading level in validation studies. The SMOG formula is based on the number of words consisting of three or more syllables. The SMOG score is commonly used to assess readability of healthcare information.
Tips for writing readable Consent Forms:
- PRISM online training is a web-based plain language tutorial for research professionals, including scientists, research staff, Institutional Review Boards (IRBs), or communications staff. The hour-long training covers health literacy and readability, plain language strategies and examples, and interactive editing examples and exercises.
- There are a variety of tools available to assist investigators in improving readability of CFs. PRISM online training addresses some tactics. The PRISM Readability Toolkit is available here.
- The Readability Toolkit is a plain language handbook illustrating how to improve the readability of research consent forms and other materials for study participants. The Toolkit includes plain language principles and strategies, quick reference guide and editing checklist, Plain language alternatives to complex terms, easy-to-read template language for consent forms and links to readability resources.
| Do | Do Not |
|---|---|
| Use short sentences and use words familiar to the non-medical reader. | Use medical terminology without explaining it, or use words that an 8th grader would not understand. |
| Refer to thesauruses and medical glossaries made for children to find alternative ways to refer to medical terminology. | Use medical “jargon” or words longer than three syllables, when another word is also appropriate. |
| Use the second person (“you”) and note that they are asked to participate in a research study. Be personal. | Use the third person (“the subject”), and avoid writing “invite” to refer to their participation. |
| Use pictures and graphs wherever possible. | Provide information solely in large blocks of text, with long sentences. |
| Say “for example” or “so forth.” | Say e.g. or etc. |
| Use tablespoons or teaspoons to refer to the measurement of bodily fluids, and spell them out. | Use ml or cc to refer to volumes of bodily fluids, or abbreviate teaspoons/tablespoons. |
| Say “greater than” or “less than”. | Use “>” or “<” or other symbols that an 8th grader might have trouble understanding at first glance. |
| Describe study terminology such as “randomized”, “placebo”, or “double blind”; such as “like the flip of a coin”. | Use medical or study terminology without explaining it in lay terms. |
| Use the words “study drug” or “study regimen”. | Use the terms “therapy” or “treatment” to describe drugs, devices, or procedures. |
| Refer to investigational drugs or devices as “experimental” or “investigational”, and that it means the FDA has not yet approved it. | Refer to investigational drugs or devices as “new.” |
Readability Examples and Templates:
- Invitation and Summary Template
- Invitation and Summary Sample 1 (Phase I Oncology Drug)
- Invitation and Summary Sample 2 (QOL Oncology)
- Invitation and Summary Sample 3 (Phase III Drug)
- Invitation and Summary Sample 4 (Knee Replacement)
Consent Teach-Back Tool
UNMC is recommending that study teams utilize the "teach-back" method during the informed consent process for their clinical studies. This method will enhance the informed consent process, ensuring participants are well-prepared before consenting and enrolling in clinical studies.
Our aim is to empower clinical research investigators and coordinators with the tools and knowledge to ensure that research study participants are fully informed and understand all aspects of the informed consent form.
Ethical Responsibility in Research
- Disclosure: It's our duty to provide all necessary information to potential research participants.
- Decision-making: We must ensure that participants can make informed decisions based on the information provided.
The Importance of Health Literacy
"Health literacy plays a crucial role in maintaining a healthy lifestyle and making informed decisions about our health care. Yet, a report from the HHS Surgeon General in 2019 highlighted that only 12% of Americans possess proficient health literacy skills. This underscores the importance of clear communication in the informed consent process."
Reference: HHS Surgeon General Reports and Publications, 2019
Consent Teach Back: A Strategy for Clarity
- A strategy to improve the researcher’s ability to explain the ICD content in a way that is clear and understandable.
- An opportunity to facilitate understanding of why a participant may or may not want to participate.
- A tool to assure that the participant is provided with sufficient opportunity to discuss the information given to them and to consider whether or not to participate
The 5 T’s of Teach Back
- Triage: Concentrate on one topic at a time.
- Tools: Utilize models, written tools, posters, graphics, etc., to aid in explaining the desired information.
- Take Responsibility: Phrase it as, “I want to make sure I did a good job explaining…”
- Tell Me: Encourage the participant to express their understanding in their own words. Be specific about what you expect them to relay back.
- Try Again: If the participant's understanding isn't clear, revisit the topic.
Reference: Anderson, Leister and DeRego, Health Literacy Research and Practice, 2020
Evaluating Understanding with Teach-Back
- Participant’s Understanding of the Study:
- Purpose
- Procedures
- Risks
- Appreciating the Consequences of Participation:
- Recognizing it's a research study
- Potential benefits (or lack thereof)
- Impact on current or future treatments
- Confidentiality and data access
- Participant’s Reasoning/Decision Process:
- Awareness of other options
- Understanding that participation is VOLUNTARY
- Participant’s Ability to Make a Choice.
E-Signature Instructions
The e-signature function in RSS is a way to electronically sign consent forms. It can be used if the consent process is conducted in person (face to face) or remotely (telephone or video as approved by the IRB). RSS e-signature can only be used for certain studies, outlined in the table below.
E-signature can only be used on newly created Narrative Consent Forms.
When can RSS e-signature be used? (as of 2/20/2024)
| Study Types | |
|---|---|
| FDA-regulated (drug/device) studies | Yes |
| Non-FDA-regulated studies | Yes |
| Commercially funded studies | Yes |
| Federally funded studies | Yes |
| CIRB (studies relying on an external IRB) | No |
| Multi-site studies where UNMC is the IRB of record | No |
Consent forms that are electronically signed in RSS are maintained in the RSS application, so paper forms do not need to be printed and stored. See the links below for instructions on in person or remote e-signatures:
If you have any questions or would like to schedule a training session, please contact Sue Logsdon at slogsdon@unmc.edu.
IRB Training Videos
Below are training topics related to the IRB, its history, and its function. Please let these videos serve as a training tool for those new to the IRB and research fields. If you need individual assistance with specific issues, or for general information, please contact us at irbora@unmc.edu or through the RSS Message Portal.
Basics: Introduction to the Institutional Review Board (2021)
Basics: New to Human Subjects Research? (5/5/2025)
Ethical Access to Patients as Human Subjects of Research (2021)
Getting Approved by the IRB: Everything You Need to Know (2021)
Human Subject Research vs Not Human Subject Research (5/8/2025)
Informed Consent (2021)
IRB Considerations in Research Involving Drugs and Devices (2021)
Use of the IRB Short Form (2021)
RSS Training
Guides to assist with RSS procedures:
If there is a topic that does not appear below, please contact irbora@unmc.edu
Adding Personnel
Adding Documents to an Application
Consent Forms 101
Creating and navigating a New Application
Deleting Personnel
Functions within a Consent Form
How to submit an Incident Report
Navigating the RSS Dashboard
Signatures on a Consent Form
Submitting a Change Request
Using the in-person RSS E-Signature
Using the remote RSS E-Signature
Working within an Application 101
Virtual Training/Office Hours
IRB Office Hours
We can be reached by phone (402-559-6463) or by email (irbora@unmc.edu) Mon-Fri 7am-5pm.
The IRB offers two virtual learning/training sessions each month.
Please direct questions regarding virtual training hours to the IRB Education Coordinator: Megan Berger mberger@unmc.edu or 402-559-6044
Forms
The following sections (located in the menu below will assist researchers by providing various tools and resources.
The IRB uses an electronic IRB submission system referred to as RSS. Never used the system before? Check the RSS Training section to learn more.
The IRB's Policy & Procedures Manual is available in UNMC Guides - https://guides.unmc.edu/books/hrpp-policies-and-procedures
Existing paper protocols
NOTE: THE PAPER FORMS ON THIS PAGE ARE ONLY TO BE USED FOR STUDIES THAT HAVE NOT BEEN TRANSFERRED TO THE ELECTRONIC SYSTEM (I.E. THEY ARE STILL IN PAPER FORMAT).
FORMS FOR EXISTING PAPER PROTOCOLS ONLY
| Title | Version | Format |
|---|---|---|
| 1. CR Biomedical Research | 06/13/2022 | Word |
| 2. CR Social and Behavioral Research | 06/13/2022 | Word |
| 3. CR Human Biological Materials | 06/13/2022 | Word |
| 4. Study Completion Report | 07/27/2010 | Word |
| 5. Request for Change in Protocol (Depending on the nature of the change completion of a protocol addendum may be required. Please contact the ORA for details.) | 03/10/2014 | Word |
| 6. Request for Change* (Ads, Educational Items and Personnel Only) | 05/14/2013 | Word |
| 7. Additional Personnel Change Form Pages (if more than four spaces needed) | 08/17/2010 | Word |
| 8. P&T Drug Registry Form | 04/14/2022 | Word |
FWAs - Federal Wide Assurance
The UNMC IRB Federal Wide Assurance documents can be found here:
Adult IRB Roster -2025 (FWA00002939)
Pediatric IRB Roster -2025 (FWA00002939)
Miscellaneous
| Title | Version | Format |
|---|---|---|
| Investigator Assessment Checklist for Regulatory Documentation | 9/17/2007 | Word |
| Investigator Assessment of IRB | 9/7/2007 | Word |
Recruitment templates
HRPP policy to review: #3.5 Subject Recruitment Through Advertisements
| Recruitment Type | Format |
|---|---|
| Flier - full page | Word |
| Ad - one column | Word |
| Ad - two column | Word |
| Ad - six inch | Word |
Short Forms
A Short Form may be used when a subject/LAR who cannot understand English is unexpectedly encountered, there is not sufficient time to develop and obtain IRB approval for a complete ICF written in language understandable to the subject/LAR, AND the research presents the prospect of direct therapeutic benefit to the subject.
Use of the Short Form is limited to enrollment of no more than three subjects per language in a given protocol. In order to enroll more than three subjects, the PI is required to have the complete ICF translated into the appropriate language and reviewed and approved by the IRB.
The investigator must complete a Short Form Request (available in RSS) for each subject, and submit to the ORA. Each request must be approved before the Short Form may be used.
Depending on the nature and duration of the research, the IRB may determine that the English version of the complete ICF must be translated into a language understandable to the subject and a copy given to the subject as soon as possible after enrollment. In general, this may be required for studies which are significant risk and of long duration.
Please refer to HRPP Policy 5.5 (Use of the Short Form Consent Document) for detailed instructions.
English translations of these short forms is included at the bottom of the page. Translated Short Forms generously supplied by Office of Human Subjects Research - Johns Hopkins Medicine. (Key: * - Johns Hopkins University)
Short Forms
Translated Short Forms generously supplied by Office of Human Subjects Research - Johns Hopkins Medicine (Key: * - Johns Hopkins University)
Subject's rights & responsibilities
Research Questions:
Amheric - Research Questions & Rights and Responsibilities
Rights of Research Subjects:
Rights and What to know:
Interpretation Certificates:
Single & Central IRB
For applications with due dates on or after Jan 25, 2018, and contract solicitations published on or after Jan 25, 2018, NIH expects that all sites participating in multi-site studies, which involve non-exempt Human Subjects Research(HSR) funded by the NIH, will use a single Institutional Review Board(sIRB) to conduct the ethical review required for the protection of human subjects.
This policy applies to the domestic sites of NIH-funded multi-site studies where each site will conduct the same protocol involving non-exempt HSR. It does not apply to career development, research training or fellowship awards. Implementation of the NIH sIRB policy is expected to reduce unnecessary administrative burdens and systemic inefficiencies while maintaining appropriate human subjects protections.
_________________________________________________________________________________________________________
The UNMC sIRB team serves as resource for UNMC and external site study teams collaborating on multi-site projects.
Our mission is to provide high quality, timely IRB review for multi-site HSR.
Please speak with an sIRB analyst prior to submitting an sIRB request. Any questions regarding applications, fee schedules, consultations, and more please contact the sIRB team at sirb@unmc.edu.
_________________________________________________________________________________________________________
For multi-center studies, the central IRB (cIRB) is the IRB that conducts reviews on behalf of all study sites that agree to participate in the centralized review process. For sites at institutions that have an IRB that would ordinarily review research conducted at the site, the cIRB should reach agreement with the individual institutions participating in centralized review and those institutions' IRBs about how to apportion the review responsibilities between local IRBs and the cIRB (21 CFR 56.114).
Glossary
Cede Review
An institution agrees to transfer IRB review and oversight authority for specified research to another institution’s IRB (reviewing IRB).
cIRB
<<<< insert definition >>>
Lead Site
<<<< insert definition >>>
Lead Principal Investigator (Lead Site PI)
The study wide lead Principal Investigator with ultimate responsibility for the conduct and integrity of multisite research.
Local Context
Unique legal requirements, cultural or religious values, or other site-specific variables that exist at a site where subjects are enrolled in research.
Participating Site (pSite)
<<<< insert definition >>>
Participating Site Principal Investigator (pSite PI)
The lead investigator at each institution participating in multisite research usually responsible for the conduct of the research at the participating institution.
Reliance
<<<< insert definition >>>
Reliance Agreement (also known as an Authorization Agreement)
An agreement between two Organizations engaged in human subject research that documents respective authorities, roles, responsibilities, and communication between the reviewing and relying IRBs.
Reliance Negotiation
<<<< insert definition >>>
Relying Institution
A participating Institution that cedes IRB review to the IRB of record (reviewing IRB) designated under a Reliance Agreement.
Relying IRB
<<<< insert definition >>>
Reviewing IRB
The IRB which is responsible for conducting IRB review and approval as described in 45 CFR 46.109 for cooperative human subject research.
sIRB
<<<< insert definition >>>
sIRB - What is Single IRB?
Single IRB is a model of IRB review that is intended to streamline and reduce administrative burden on IRBs reviewing multisite studies. In a single IRB model, one IRB serves as the reviewing IRB or “IRB of Record” and the participating site (pSite) IRBs serve as relying IRBs. The responsibilities of each IRB are outlined in a document called a reliance agreement.
For more information on the history of single IRB (sIRB) review and sIRB review at UNMC, see each section below:
Single Site Research:
Below is a model of single site research, or research that is only taking place at one site.
Multi-site Research:
Historically, multisite research was overseen by multiple independent IRBs. This often led to differences in IRB review and outcomes across sites.
Below is a model of historical multisite research, or research that took place at more than one site.
Single IRB Review Requirement:
The requirement for single IRB review originated from the NIH single IRB policy and the Revised Common Rule.
From the NIH:
"expectation that all [domestic] sites participating in multi-site studies involving non-exempt human subjects research funded by the National Institutes of Health will use a single Institutional Review Board (sIRB)" (NOT-OD-16-094, June 2016)
- effective date January 25, 2018
For more information on the NIH Single IRB Policy visit: https://grants.nih.gov/policy/humansubjects/single-irb-policy-multi-site-research.htm.
For Guidance on exceptions to the NIH Single IRB Policy, visit: https://grants.nih.gov/grants/guide/notice-files/NOT-OD-18-003.html
From the Revised Common Rule:
"Any institution located in the United States that is engaged in cooperative research must rely upon approval by a single IRB …" (§_.114(b)(1))
- effective date January 20, 2020
For more information on the Revised Common Rule's Single IRB Requirement, visit: https://www.ecfr.gov/current/title-45/subtitle-A/subchapter-A/part-46#46.114.
For a list of Common Rule Departments and Agencies, visit: https://www.hhs.gov/ohrp/regulations-and-policy/regulations/common-rule/index.html
For information on Common Rule Exception Determinations, visit: https://www.hhs.gov/ohrp/regulations-and-policy/single-irb-requirement/index.html#exceptions
In short, this means that studies that are federally funded and taking place at more than one site are now required to use a single IRB as the IRB of record. There are many circumstances where it is not clear if a specific study requires single IRB review or if an exception may apply. In these cases, please email sirb@unmc.edu for assistance at the earliest opportunity.
Note: Participating site IRBs are still involved in the single IRB process and have their own local requirements, but the responsibility for the regulatory component of review is ceded to the single IRB. Each IRB’s responsibilities are outlined in a document called a Reliance Agreement. More information on reliance can be found in the sIRB Reliance section below.
Single IRB Review Models:
Some groups were already utilizing a single IRB model prior to the requirement (e.g., commercial IRBs).
Below is a simplified model of how some commercial IRBs handle multisite research. Many will interact directly with the sponsor, perform IRB review activities, and then push approvals to all sites simultaneously.
Below is another model of how a single IRB may handle multisite research. In this example, the sponsor works with each site PI and study team independently. Each site submits their own application to the single IRB for review and receives separate approvals.
sIRB Review at UNMC:
Single IRB Review at UNMC is broken out into two main parts. First, the overall study and lead site-specific requirements are reviewed. After overall study and lead site approval, participating sites (pSites) are onboarded and approved on a rolling basis.
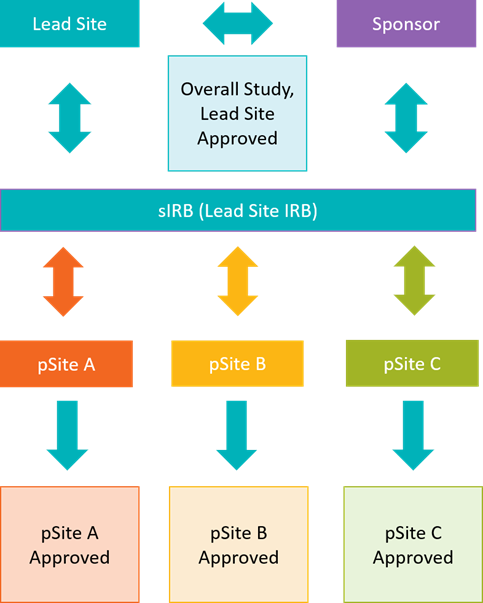
Note: pSite IRB processes are not included in the above models. Please contact your pSite IRB directly with questions.
sIRB - UNMC process
The UNMC sIRB Process is captured in the diagram below.
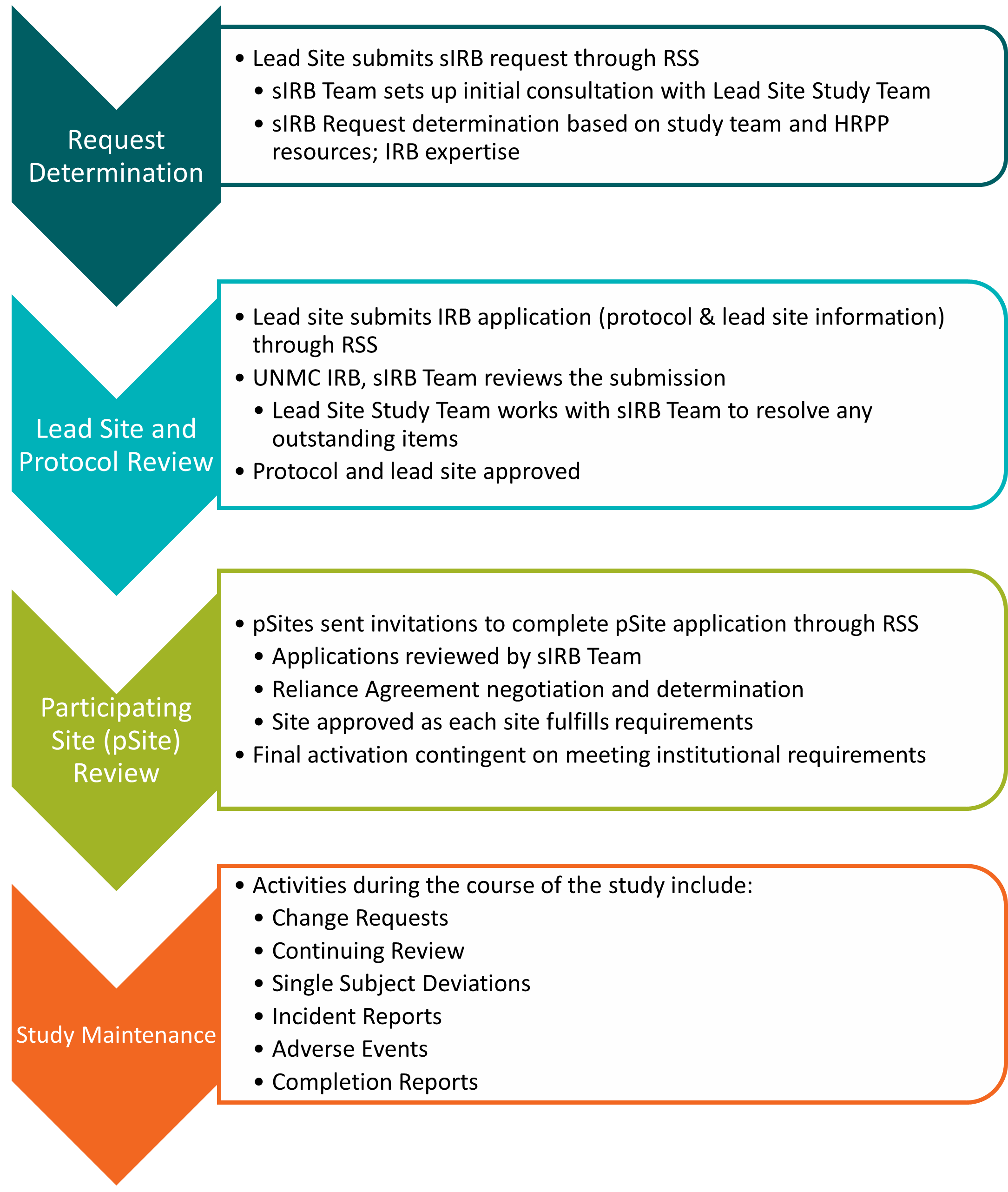
Contact sirb@unmc.edu with any UNMC sIRB process questions.
sIRB - Submission deadlines
sIRB Submission Deadlines for New Submissions, Previously Tabled Protocols and Requests for Change
- Deadline for Continuing Review submission is the 1st day of the month prior to expiration.
- Full Board Deadlines (printable PDF)
sIRB Full Board Submission Deadline – 9 am
| Submission Deadline | Date of sIRB Meeting |
|---|---|
| 2/6/2025 | 2/14/2025 |
| 3/6/2025 | 3/14/2025 |
| 4/3/2025 | 4/11/2025 |
| 5/1/2025 | 5/9/2025 |
| 6/5/2025 | 6/13/2025 |
| 7/3/2025 | 7/11/2025 |
| 7/31/2025 | 8/8/2025 |
| 9/4/2025 | 9/12/2025 |
| 10/2/2025 | 10/10/2025 |
| 11/6/2025 | 11/14/2025 |
| 12/4/2025 | 12/12/2025 |
| 12/31/2025 | 1/9/2026 |
sIRB - Reliance
Reliance refers to an arrangement where an IRB allows another IRB to perform review and approval of a study.
The UNMC IRB uses the term “single IRB” or “sIRB” to indicate that UNMC is serving as the reviewing IRB (i.e., the IRB of record) of a multisite study.
Reliance Agreement
A reliance agreement is a formal, written document that provides a mechanism for an institution engaged in research to delegate IRB review to another IRB. This document generally outlines the responsibility of the single IRB and the participating site. Often the reliance agreement is accompanied by addenda or other forms that are designed to further clarify responsibilities or collect pSite local context information.
Reliance agreements come in many formats. They may also be called:
UNMC's Preferred Reliance Agreement Format
UNMC is a SMART IRB participating site and strongly prefers to document reliance using the SMART IRB Online Reliance System (SMART IRB ORS). In addition, we ask that sites complete the SMART IRB Implementation Checklist. This will be provided during the reliance negotiation.
Note: SMART IRB is not an IRB, but a tool that IRBs use to document reliance.
Note: The UNMC sIRB Team has elected to initiate and maintain requests in the SMART IRB ORS, rather than have the lead site investigator or study team create requests. Please contact sirb@unmc.edu with questions.
If a site is not a SMART IRB participating institution, other reliance formats will be made available for use during the reliance negotiation.
Reliance Negotiation
The term reliance negotiation refers to the process of the reviewing and relying IRB representatives communicating and agreeing to the reliance agreement format and terms. The UNMC sIRB Team will initiate the negotiation during the pSite Onboarding stage of review.
sIRB - Fees
For information regarding the fee schedule for Single IRB studies, please contact sirb@unmc.edu.
cIRB - What is Central IRB?
The UNMC IRB uses the term “central IRB” or “cIRB” to indicate that the reviewing IRB (i.e., the IRB of record) of a multisite study is external to UNMC.
The UNMC IRB has contracted with two commercial IRBs, Advarra and WCG. In addition, the UNMC IRB partners with consortium, academic, and medical center IRBs nationwide.
For more details around the use of a cIRB, including when the use of an external central IRB is not permitted, see UNMC HRPP Policy 1.4 (UNMC Ceding Review to an External Central IRB).
cIRB - UNMC Process
Note: When requesting cIRB review, study teams must satisfy the requirements of each IRB and the funding agency or sponsor (as applicable), prior to initiating the research.
UNMC cIRB Initial Review Process
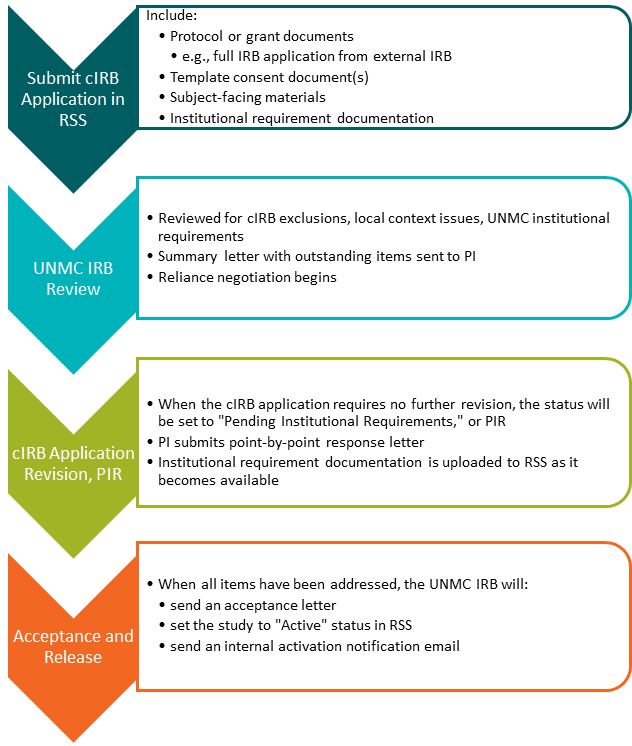
External cIRB Process
- Contact your external cIRB representative for details. Each IRB operates differently.
- IRB approval is needed prior to initiating research
Funding Agency/ Sponsor Process
- Contact your funding agency/sponsor representative for details. Each group operates differently.
- Funding agency/sponsor approval is needed prior to initiating research
Post-acceptance Study Activities
In general, the external IRB of record will be responsible for reviewing most study activities. In some cases, these may also need to be submitted to the UNMC IRB.
Things to submit to the external cIRB:
- Amendments
- Continuing Reviews
- Single-subject Deviation Requests
- Short-form Requests
- Incident Reports
- Adverse Event Reports
- Study Completion Reports
Note: In most cases, submit to the external IRB. If determined to be necessary, the external cIRB will notify the UNMC IRB directly or request the study team notify the UNMC IRB.
Things to submit to the UNMC IRB:
- New or modified Conflicts of Interest, management plans or additional external cIRB requirements
- Copies of reports made to OHRP and/or FDA
- Copies of Incident Reports submitted to the cIRB
- Copies of Internal Adverse Event Reports submitted to the cIRB
- Personnel Changes
Note: personnel are NOT permitted to work on a study until UNMC IRB approval is received.
- Annual Status Update via Demographic Recruiting Numbers Form
- Study Closure Notifications
Note: Send a notification in the message portal, indicating the study is now closed. Upload applicable documentation.
cIRB - Reliance
When requesting to rely on an external IRB, the UNMC IRB will assess the external IRB’s qualifications in accordance with HRPP Policy 1.4 (UNMC Ceding Review to an External Central IRB). The UNMC IRB may request information related, but not limited, to:
- SMART IRB Participation
- HRPP Accreditation or Completion of OHRP QA Self-Assessment Tool
- Valid FWA, Registration with OHRP and FDA (as applicable)
The UNMC IRB strongly prefers to document reliance using the SMART IRB Online Reliance System. UNMC has several reliance templates based on institution type available for use. Other reliance formats are approved on a case-by-case basis.
cIRB - Fees
In line with most other academic medical centers and universities, the Office of Regulatory Affairs and the IRB charges an initial submission and an annual continuing review fee for commercial and industry sponsored protocols.
Central IRB:
| effective 7/1/2021 | Initial Submission | Annual Continuing Reviews |
|---|---|---|
| Central IRB Review | $3000 | N/A |
Commercial/Industry Sponsored Research Protocols:
| effective 7/1/2021 | Initial Submission | Annual Continuing Reviews |
|---|---|---|
| Full Board | $3000 | $1250 |
| Expedited | $2000 | $1000 |
Center for Nutrition & Health Impact
| Please contact sirb@unmc.edu for more information. |
|---|
Single IRB:
| Please contact sirb@unmc.edu for more information. |
|---|
cIRB - Forms & Links
Note: the UNMC IRB cannot answer questions related to external IRB processes or applications. Contact the external IRB's helpdesk or designated representative directly for assistance.
For questions regarding UNMC cIRB processes or applications, please contact sirb@unmc.edu.
Advarra
- Advarra IRB Application System, CIRBI: https://www.cirbi.net
- Approved UNMC - Advarra Consent Local Context Information Document (11/2/2023)
- Advarra Checklist (optional)
Advarra has requested the UNMC IRB number be added to the application as follows:
- For single-site protocol applications: in the field below in the screen shot (with the x’s).
- For site only (SSU) applications: add the internal number to the following area on the investigator application:
Contact Information Sheet
The UNMC IRB recognizes that not all central IRB consent templates have the option to add local study contacts. The cIRB application in RSS now has the option to build a “Contact Information Sheet” to be provided to subjects along with the approved consent documents.
***Older studies within RSS may not have this functionality. The Contact Information Sheet Template may be downloaded for manual completion.
Miscellaneous central IRBs
- Contact the external IRB for information regarding their IRB review process and any applicable IRB application management systems.
- UNMC cIRB Local Context Information Document (11/2/2023)
NCI CIRB
- NCI CIRB Application System, IRBManager: https://nci.my.irbmanager.com/
- Approved Univ of Nebraska Consent Local Context Information Document (9/7/2022)
NMDP
SMART IRB
- SMART IRB https://smartirb.org/ is not an IRB, but a tool used to document reliance between IRBs.
- The IRB representatives from each institution will manage this negotiation. Unless otherwise indicated, nothing is needed from the investigator or study teams to fulfill this requirement.
WCG
- WCG IRB Application System, WCG IRB Connexus: https://identity-connexus.wcgirb.com
- Approved U of Nebraska - WCG Consent Local Context Information Document (1/12/2024)
If requesting revisions to the template consent language, the University of Nebraska – WCG ICF Checklist must be completed and signed by the study’s lead UNMC IRB representative.
SROC
In accordance with all Federal Guidelines, the University of Nebraska and Board of Regents policy, the Scientific Research Oversight Committee (SROC) must review and approve all research using hESC lines and ONLY lines from the NIH Human Embryonic Stem Cell Registry (http://grants.nih.gov/stem_cells/registry/current.htm?sort=rnd) are allowed. Cell lines which are not on this registry are not allowed to be used for NU System research. The NIH Registration Number for a proposed stem cell line must be cited on your application for hESC use when submitting your application to the SROC.
The committee must be composed of scientists who have hESC expertise and at least 2 non-affiliated members, one of which must be a non-scientist.
Human embryonic stem cell research at UNMC:
Current and prior protocols address a wide range of topics from the very basic mechanisms that regulate stem cell self renewal, which is the primary characteristic that distinguishes stem cells from all other cell types in the body, to potential applications of stem cells in regenerative medicine.
Studies have demonstrated the derivation of human hepatocytes (liver cells) from hES cells, retinal and neuronal (eye and brain) cells and shown that stem cells that are differentiated to fibroblasts of the lung in conditions similar to those in patients with asthma, are much more contractile than normally derived lung fibroblasts. This has prompted a new hypothesis to explain the persistence of asthma even in the face of anti-inflammatory treatments. Additional projects address the education aspects of the properties of hES cells. These results have been published in nationally ranked and recognized medical journals.
All studies must be SROC and IRB approved and employ federally approved hES cell lines that are used according to all federal, state and university regulations.
Chairs and Administrator

Iqbal Ahmad, PhD
- Chair, SROC
- 402-559-4091

- IRB Analyst III, SROC
- 402-559-3779
Committee
In 2008, the Human Embryonic Stem Cell Research Advisory Committee of the National Research Council and Institute of Medicine published Amendments to the National Academies’ Guidelines for Human Embryonic Stem Cell Research.
Section 2.0 (page 11) describes the establishment of an Institutional ESCRO “To provide oversight of all issues related to derivation and use of hES (human embryonic stem) cell lines and to facilitate education of investigators involved in hES cell research, each institution should have activities involving hES cells overseen by an Embryonic Stem Cell Research Committee (ESCRO).”
“An ESCRO committee should include independent representatives of the lay public as well as persons with expertise in developmental biology, stem cell research, molecular biology, assisted reproduction and ethical and legal issues in hES cell research.” (Since in Nebraska, development of human stem cell lines is not permitted, assisted reproduction input is potentially less relevant than in states that do not have this prohibition.)
“It must have suitable scientific, medical and ethical expertise to conduct its own review and should have the resources needed to coordinate the management of various other reviews required for a particular protocol.” (eg animal use, recombinant DNA, human subject research)
“But the ESCRO committee should not be a subcommittee of the IRB, as its responsibilities extend beyond human subject research.” (For example, in Nebraska, this includes ensuring that all hES cell lines employed are federally approved).
“The ESCRO Committee should
- Provide oversight over all issues related to deviation and use of hES cell lines.
- Review and approve the scientific merit of research protocols.
- Review compliance of all in-house hES cell research with all relevant regulations and these guidelines.
- Maintain registries of hES cell research conducted at the institution and hES cell lines derived (not relevant in Nebraska) or imported by institutional investigators.
- Facilitate education of investigators involved in hES cell research.”
The UNMC Scientific Research Oversight Committee (SROC) undertakes all of these tasks and reports its actions to the UNMC IRB and the Chancellor.
Forms
IRB Staff
This section will detail contact information for both IRB Staff and IRB Chairs.
IRB Analysts/Staff
| Deron Anderson, MS |
|---|
| IRB Analyst |
| 402-559-5845 |
| Megan Berger, BS, MEd |
|---|
| IRB Analyst & Education Coordinator |
| 402-559-6044 |
| Erika Boohar, BA |
|---|
| IRB Analyst |
| 402-559-3853 |
| Bobbi Chapman, MS |
|---|
| IRB Analyst |
| 402-559-6119 |
| Kristi DeHaai, MS, CIP |
|---|
| IRB Analyst |
| 402-559-9991 |
| Emily Gale, BS |
|---|
| IRB Analyst |
| 402-559-7269 |
| Bruce Gordon, MD Biography |
|---|
| Assistant Vice Chancellor for Regulatory Affairs |
| Executive Chair, Institutional Review Boards |
| Professor, Division of Pediatric Hematology/Oncology |
| 402-559-6463 |
| Gail D. Kotulak, BS, CIP |
|---|
| IRB Analyst |
| 402-559-6540 |
| Sue M. Logsdon, MS, CIP |
|---|
| IRB Analyst |
| 402-559-3779 |
| Bryan Ludwig, BA, CIP |
|---|
| IRB Analyst |
| 402-559-8561 |
| Jessica Odvody-Wernimont, BS |
|---|
| IRB Analyst |
| 402-559-4741 |
| Nancy Olson, JD |
|---|
| IRB Analyst |
| 601-946-9020 |
| Karli Storm-Narvainen, PhD |
|---|
| IRB Analyst |
| 402-559-6898 |
| Nicole Weber, BA |
|---|
| IRB Analyst |
| 402-559-6596 |
IRB Chairs
The Institutional Review Board (IRB) is composed of members from a variety of scientific disciplines and individuals from the community who assist investigators in the protection of the rights and welfare of human subjects.
Executive Chair

Bruce Gordon, MD
Executive Chair, UNMC Institutional Review Board
Chair, Joint Pediatric Institutional Review Board
402-559-6463
Chairs

Elizabeth Beam, PhD, RN
Vice Chair, IRB-01 | Vice Chair, IRB-05
UNMC Institutional Review Board
402-559-6547

Matthew Lunning, DO
Vice Chair, IRB-01 | Vice Chair, IRB-05
UNMC Institutional Review Board
402-559-7164

Andrew Macfadyen, MD
Vice Chair, Children's Nebraska Joint Institutional Review Board
UNMC Institutional Review Board
402-955-4235

Suzanne Nuss, RN, MBA, PhD
Vice Chair, IRB-03 | Chair, IRB-05
UNMC Institutional Review Board
402-559-8815

Sheilah Snyder, MD
Chair, Children's Nebraska Joint Institutional Review Board
UNMC Institutional Review Board
402-955-8125

Douglas A. Stoller, MD, PhD
Vice Chair, IRB-01
UNMC Institutional Review Board
402-559-1350

Benjamin Teply, MD
Vice Chair, IRB-02
UNMC Institutional Review Board
402-559-7915

Brigette Vaughan, APRN
Chair, IRB-02
UNMC/Children's Nebraska Joint Institutional Review Board
402-559-9800

Wes Zeger, DO
Vice Chair, IRB-03
UNMC Institutional Review Board
402-559-6841